


Reaction mechanisms, key challenges, and future prospects of zinc-ion/magnesium-based batteries: from fundamental researches to electrode/electrolyte engineering
 0
0 Abstract
Greenhouse gas emissions from fossil fuel combustion accelerate global climate change, while the inherent intermittency of wind and solar renewables demands advanced energy storage systems to support their large-scale grid integration. Lithium-ion batteries (LIBs) currently prevail in the field of energy storage, yet their extensive commercial deployment is hindered by geographically uneven lithium reserves, steeply rising raw material prices and intrinsic safety risks. In this context, zinc-ion batteries (ZIBs) and magnesium-ion batteries (MIBs) have emerged as highly promising multivalent metal-ion alternatives. Benefiting from abundant reserves, cost-effectiveness, high theoretical capacities, and superior intrinsic safety, they represent formidable contenders to succeed LIBs. To provide a comparative and engineering-centric perspective, this review systematically evaluates these two multivalent systems within a unified framework encompassing charge storage mechanisms, material-level challenges, interfacial engineering, and device integration. By elucidating their common technical bottlenecks and divergent solutions, this review intends to deliver cross-system research insights, identify transferable strategies, and outline a clear development roadmap spanning from fundamental investigation to practical industrial application, this work thus acts as a strategic guideline for researchers and engineers engaged in multivalent metal-ion batteries.
Keywords
INTRODUCTION
The combustion of fossil fuels constitutes the predominant source of anthropogenic carbon dioxide (CO2) emissions, driving the unprecedented accumulation of atmospheric greenhouse gases. This persistent accumulation is widely recognized as the principal driver of global climate change[1]. In response to the environmental imperatives associated with fossil fuel depletion and climate change, transitioning to clean, and renewable energy has emerged as a global consensus. Consequently, decarbonization and strategic emission mitigations represent the primary pathways toward achieving this sustainable transformation[2]. Although the penetration of solar and wind energy into the global power grid is expanding rapidly, these renewable sources are inherently characterized by intermittency and volatility. Consequently, the development of highly efficient energy storage systems (ESS) is imperative to balance power supply and demand during periods of low generation[3,4]. Due to their high energy density, extended cycle life, and mature industrial supply chain, LIBs have emerged as the dominant technology in electric vehicles and consumer electronics, while also seeing widespread deployment in large-scale energy storage systems[5-9]. Global demand for lithium is surging at an unprecedented pace. Stimulated by the concurrent expansion of electric vehicles and grid-scale energy storage, the International Energy Agency (IEA) forecasts that lithium demand could escalate 30-fold by 2030 and potentially exceed 100-fold by 2050. However, inherent safety liabilities, fluctuating market expenses, and the limited, unevenly distributed reserves of lithium impose severe bottlenecks on the further large-scale deployment of Lithium-ion batteries (LIBs)[10-12].
As the global demand for renewable energy storage intensifies, conventional lithium-ion batteries struggle to fully satisfy the requirements of increasingly diverse application scenarios. Consequently, alternative battery technologies have attracted growing attention as viable pathways toward more efficient and eco-friendly energy storage. Among these candidates, Zinc-based batteries stand out for large-scale energy storage applications owing to their abundant resources, cost-effectiveness, environmental benignity, and exceptional safety profiles. These prominent advantages are inherently underpinned by the high theoretical capacity of the Zn anode (820 mAh g-1), its low cost, non-toxic nature, and a moderate redox potential[13-16]. Compared with LIBs, zinc-ion batteries (ZIBs) demonstrate significant advantages in both cost and security[14,17-24]. The crustal abundance of zinc (~70 ppm) substantially surpasses that of lithium (~20 ppm) and exhibits a more uniform global distribution, granting it significant advantages in resource sustainability. This geological profile is complemented by a mature, stable zinc supply chain and a well-established recycling ecosystem. In stark contrast, global lithium reserves are highly centralized, with approximately 60% located within the South American “lithium triangle”. This extreme geographic concentration renders the lithium supply highly vulnerable to geopolitical instabilities and the skyrocketing demand driven by the clean energy transition[25,26]. In particular, ZIBs employ non-flammable mild aqueous electrolytes, which greatly reduce the risk of fire and explosion compared with LIBs using organic electrolytes[27,28]. However, this intrinsic safety advantage is contingent upon standard operating conditions. Under extreme scenarios -such as severe overcharging, mechanical puncturing, or elevated temperature environments- aqueous ZIBs can still undergo localized or catastrophic failure modes. These include electrolyte leakage, host electrode structural collapse, and parasitic gas evolution, which significantly compromise both cell safety and operational lifespan[29]. Meanwhile, zinb based systems can achieve high theoretical capacity and the unique advantage of two electrons participating in electrochemical reactions. The value can reach 5,851 mAh cm-3[30]. Consequently, these compelling attributes have stimulated extensive research enthusiasm toward aqueous ZIBs. Nevertheless, despite their unique promises, the widespread commercialization and large-scale application of ZIBs remain severely hindered by several intrinsic bottlenecks. First, the structural instability of host electrode materials often leads to severe capacity degradation, thereby limiting the operational cycle life. Second, uncontrolled zinc dendrite propagation and concurrent anodic corrosion drastically degrade the Coulombic efficiency and reversibility of the zinc plating/stripping processes. Concurrently, the sluggish transport kinetics of Zn2+ ions, stemming from their strong electrostatic interactions with the host matrix, impose formidable thermodynamic barriers that impede smooth ion insertion[31,32].
Following the initial report of a rechargeable magnesium battery by the Aurbach team in 1990, magnesium-ion batteries (MIBs) have rapidly garnered widespread attention due to their unique advantages[33]. Regarding resource availability, magnesium is highly abundant in the Earth’s crust and widely distributed, thereby significantly lowering the raw material costs for battery production[34]. In terms of electrochemical performance, Mg metal anodes exhibit an exceptionally high volumetric capacity
Among the plethora of “beyond-lithium” candidates, ZIBs and MIBs have emerged as two of the most promising yet mechanistically distinct paradigms. Crucially, both systems utilize divalent charge carriers (Zn2+ and Mg2+), thereby confronting the fundamental challenges intrinsic to multivalent chemistries: strong Coulombic interactions within host lattices, sluggish solid-state diffusion, and prohibitive desolvation energy barriers[40]. ZIBs and MIBs represent two primary, contrasting strategic paradigms to address these challenges. ZIBs capitalize on the electrochemical compatibility of zinc in aqueous electrolytes, prioritizing intrinsic safety, cost-effectiveness, and high ionic conductivity, albeit constrained by the narrow stability window of water. In contrast, MIBs pursue non-aqueous (organic) formulations to unlock higher operating voltages and superior energy densities, thereby grappling with the complexities of electrolyte synthesis, severe anode passivation, and poor interfacial compatibility[41]. Their distinct development trajectories point toward highly complementary applications. ZIBs, possessing near-term viability, serve as prime candidates for grid-scale stationary energy storage where safety and capital cost are paramount. Concurrently, MIBs, characterized by superior volumetric capacities, hold long-term promise for density-critical configurations. Analyzing these systems concurrently provides a holistic perspective of the multivalent battery landscape across disparate technology readiness levels. Crucially, the primary objective of this review transcends a mere chronological cataloging of recent literature; rather, it establishes a rigorous, critical comparative analysis between ZIBs and MIBs. The unique contributions of this work are twofold: (1) A unified analytical framework is applied to both systems, isolating critical bottlenecks and systematically categorizing state-of-the-art solutions across material architecture, electrolyte engineering, and interfacial modification; (2) Priority is given to strategies designed to enhance practical performance metrics-such as operational cycle life under realistic testing conditions, cell-level energy density, and environmental adaptability - thereby moving beyond idealized laboratory-scale metrics. Through this structured, comparative approach, this review aims to distill definitive guidelines for future research, accelerating the realization of practical, high-performance multivalent-ion batteries.
Prior reviews and scope of this work
Over the past five years, the field has witnessed the publication of numerous authoritative reviews dedicating to zinc- and magnesium-based chemistries. For instance, Upreti et al.[42] centered their discussion on performance enhancement strategies for zinc metal anodes in aqueous zinc-ion batteries. Their work deeply deconvoluted the underlying mechanisms of critical bottlenecks, including zinc dendrite propagation, parasitic corrosion, and the competitive hydrogen evolution reaction (HER). Furthermore, they systematically elaborated on diverse protection schemes - ranging from fundamental electrolyte formulation to advanced “water-in-salt” configurations and deep eutectic solvents - with the ultimate goal of delivering reliable technological blueprints for grid-scale energy storage. Alawi et al.[32] focused on zinc-ion batteries, delving into the defects of this type of battery, its development opportunities, and the paths for performance optimization, providing a comprehensive reference for its application in sustainable energy storage. Wang et al.[43] systematically reviewed the current research status of zinc-ion batteries, and focused on summarizing the performance optimization methods for each component of the battery (electrodes, electrolyte, etc.), aiming to comprehensively present the technological progress and improvement directions of zinc-ion batteries. In addition, Guo et al.[44] focused on rechargeable magnesium-based batteries, concentrating on the goal of efficient energy storage. They summarized the latest research progress in this type of battery in terms of electrode materials, electrolyte systems, and battery structure design. In addition, they deeply analyzed key technical points such as the magnesium storage mechanism and performance optimization of positive electrode materials, the improvement of electrolyte compatibility and stability, and the modification strategies for magnesium-based negative electrodes. Lei et al.[45] focused on the development of high-performance rechargeable magnesium batteries, with the rational design of new energy storage systems as the core. From perspectives such as material design, structural optimization, and reaction mechanisms, they systematically explored key strategies for enhancing the performance of magnesium batteries, including rational selection and modification of electrode materials, innovative development of electrolytes, and integrated optimization of battery systems. The aim was to provide theoretical guidance and technical references for constructing rechargeable magnesium batteries with high energy density and long cycle stability.
Among the plethora of “beyond-lithium” candidates, ZIB and MIB have become the two most promising yet distinct energy storage systems. While excellent reviews exist that focus solely on the advances of Zn-ion batteries[46], or on the progress of Mg-based systems, a critical comparative analysis that examines them through the same lens is lacking. For instance, the comprehensive review by Xiao et al.[46] provides a deep dive into various Zn-based systems, establishing a solid foundation for the field. Our work distinguishes itself by building upon such foundations to offer a parallel and comparative study. The unique contributions of this review are threefold: (1) We analyze the storage mechanisms, material challenges (for both anodes and cathodes), and electrolyte engineering of the two systems from a unified perspective; (2) We specifically focus on strategies to improve practical performance metrics, such as cycle life, overall battery energy density, and extreme temperature adaptability; (3) Furthermore, we systematically compare representative cathode families, electrolyte types, and other relevant parameters between ZIBs and MIBs [Table 1]. Through this structured and comparative approach, this review aims to distill clear guidelines for future research and accelerate the development of practical, high-performance multivalent-ion batteries.
Summary of the performance of Zn-ion battery and Mg-ion battery
| Types of batteries | Cathode material | Electrolyte | Capacity (mAh g-1) | Working voltage (V) | Cycle life (cycles) | Capacity retention ratio (%) | Ref. |
| ZIBs | NH4V4O10-ZIF-67 | 2 M ZnSO4 | 399.2 | 0.2-1.6 | 1,000 | 96.6 | [47] |
| NaCa0.6V6O16·3H2O | 2 M ZnSO4 | 443.2 | 0.4-1.5 | 2,000 | 94 | [48] | |
| PbO2 | 1 M KOH + 0.1 M Zn(CH3COO)2 | 86 | 2.92 | 250 | / | [49] | |
| Co0.247V2O5·0.994H2O | / | 432 | 1.7 | 7,500 | 90.26 | [50] | |
| α-MnO2 | 1 M (NH4)2SO4 + 0.1 M MnSO4·H2O | 365 | 1.35 | 4,000 | 93.3 | [51] | |
| KFeMnHCF | 1 M ZnSO4 + 1 M KCl | 159.3 | 2.01 | 3,000 | 96.0 | [52] | |
| CoFe(CN)6 | 4 M Zn(OTf)2 | 173.4 | 1.75 | 2,200 | / | [53] | |
| VO2-N | 3 M Zn(CF3SO3)2 | 373.7 | 1.4 | 2,000 | 88 | [54] | |
| MIBs | Mo6S8 | 0.4 M APC | 124 | 0.5-2.0 | 200 | 95.9 | [55] |
| Li3V2(PO4)3 | 0.5 M Mg(ClO4)2 | 124 | 0.46 | 300 | 80 | [56] | |
| Mg0.58MnO2·0.56H2O | 0.5 M MgCl2 | 169.3 | 0-1.6 | 5,000 | 100 | [57] | |
| NixTi1-xO2-B/RGO@CNT | 0.4 M (MgPhCl)2-AlCl3 | 167.5 | 0-2.0 | 500 | 84.5 | [58] | |
| EH-MoS2 | / | 331.3 | 0.01-2.0 | 2,000 | 85.7 | [59] | |
| Mg2Ga5 | 0.4 M APC | 307.5 | 0.01-0.7 | 680 | 94 | [60] | |
| Ti3C2Tx | 0.4 M APC | 210 | 0.3-2.85 | 100 | 82 | [61] |
ZINC-BASED ENERGY STORAGE SYSTEM
Introduction of zinc-ion battery
Following the inception of the first battery, the “Voltaic Pile” in 1800, a variety of batteries have been progressively developed[62]. In the 1980s, zinc-based flow batteries emerged as a category of batteries[63]. In 2012, ZIBs were successfully developed, although their structure and performance are still being optimized today. ZIBs are a battery system featuring zinc as the anode, aqueous solutions as the electrolyte, and enabling zinc ion insertion/extraction. Zinc metal is inherently attractive due to its high crustal abundance, cost-effectiveness, and impressive theoretical volumetric capacity (5,851 mAh cm-3), which collectively provide a robust economic and electrochemical foundation for large-scale energy storage applications[64]. Additionally, aqueous zinc-based batteries exhibit an inherently high safety profile, primarily due to the employment of mild aqueous electrolytes. Furthermore, zinc and its derivatives are generally benign and exhibit low toxicity, which facilitates battery recycling, minimizes environmental impact, and aligns with current sustainability requirements.
Energy storage mechanism
Zn2+ detachment/insertion mechanism
The deintercalation of Zn2+ represents the most typical form of energy storage. This phenomenon is particularly prevalent in materials featuring layered structures (V2O5, MnO2) or tunnel structures (α-MnO2)[65]. Xu et al.[66] claim that during discharge, Zn ions embed into the tunnels of α-MnO2 to form a spinel-structured ZnMn2O4. Furthermore, through the X-ray Photoelectron Spectroscopy (XPS) and X-ray diffraction (XRD) characterization [Figure 1A and B], it was confirmed that in aqueous zinc-ion batteries, the α-MnO2 cathode exhibited a significant Zn signal at the 1.3 V insertion state and formed the ZnMn2O4 crystal phase. At the 1.7 V removal state, the Zn signal significantly weakened and the crystal phase reverted to pure α-MnO2. The original state had no Zn signal or zinc-manganese oxide phase. This clearly indicates that Zn2+ can undergo reversible insertion and removal in the tunnel structure of α-MnO2, accompanied by the reversible crystal phase transformation between α-MnO2 and ZnMn2O4. Moreover, the reaction processes of the cathode and anode are proposed:
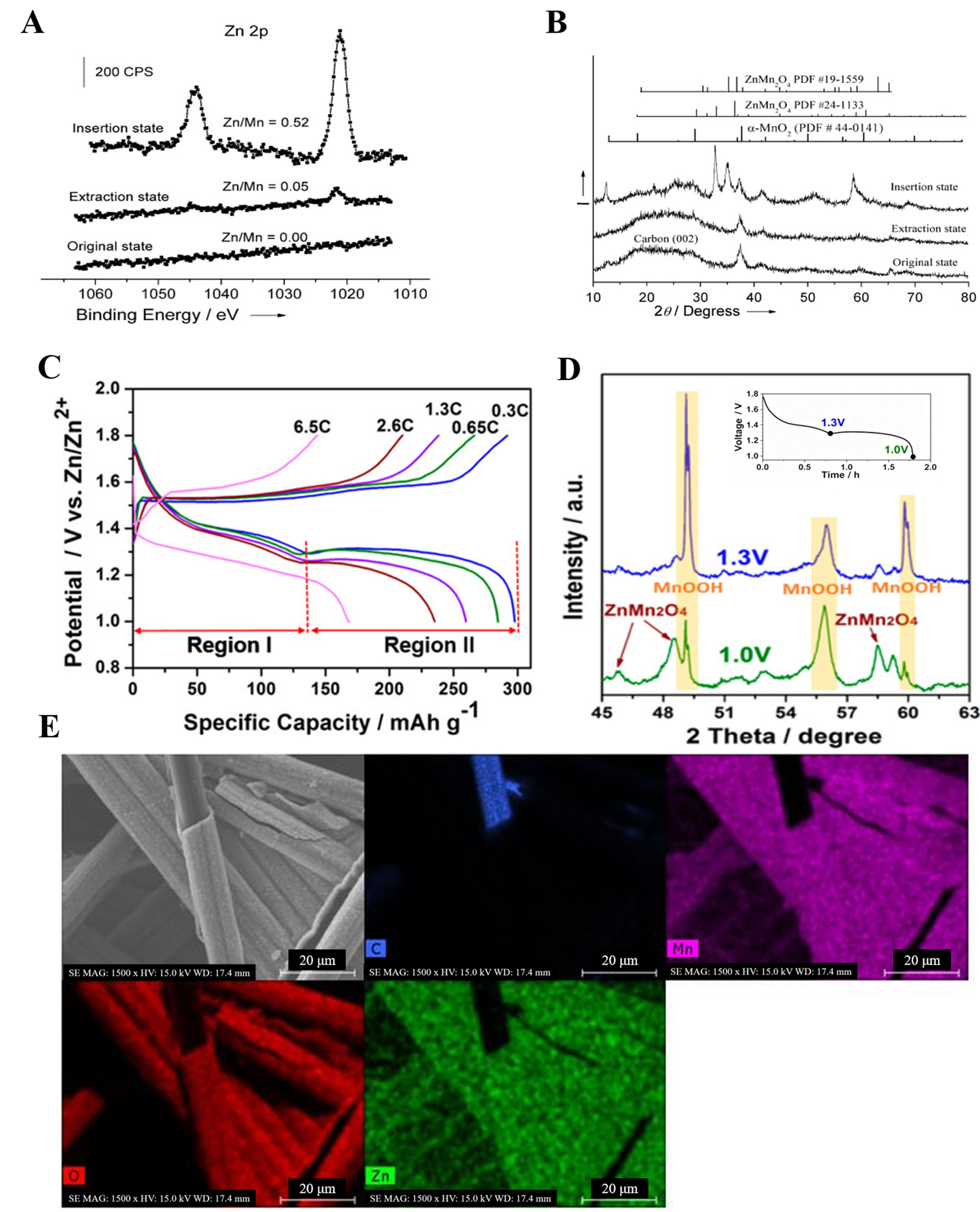
Figure 1. (A) Zn 2p core level spectra of cathodic crystalline α-MnO2 electrodes in the original, extraction, and insertion states. (B) XRD patterns of cathodic crystalline α-MnO2 electrodes in the original, extraction, and insertion states. (A and B) Reprinted with permission from[66]. Copyright 2011, Wiley; (C) charge and discharge curves at different rates in first cycle. (D) Ex-situ XRD patterns of the MnO2@CFP cathode at depth of discharge at 1.3 V and 1.0 V, respectively. (E) SEM and elemental mapping images of discharged MnO2@CFP electrode. (C-E) Reprinted with permission from[69]. Copyright 2017, American Chemical Society. XRD: X‑ray diffraction; SEM: scanning electron microscopy; CFP: carbon fiber paper.
Cathodic reaction: Zn2+ +2e- + 2α - MnO2 ↔ ZnMn2O4
Anode reaction: Zn ↔Zn2+ + 2e-
The results demonstrate that reversible transfer of Zn2+ between the anode and cathode enables energy storage and release. Zinc dissolution/deposition occurs at the anode, while Zn2+ insertion/extraction takes place at the cathode[67,68]. Moreover, high capacities can be effectively achieved through the reversible intercalation of Zn2+ into the tunnel structure of α-MnO2. Consequently, materials exhibiting tunnel, layered, or open-framework architectures are considered ideal cathodes for ZIBs, as they provide abundant interstitial sites to accommodate zinc ions.
Co-intercalation/deintercalation mechanism of H+ and Zn2+
Sun et al.[69] firstly reported on the sequential ion intercalation reaction mechanism in ZIBs. They developed a reversible Zn/MnO2 battery. The cathode was fabricated via in-situ electrodeposition on carbon fiber paper using a mildly acidic ZnSO4 + MnSO4 mixed electrolyte. They demonstrated through a series of experiments that the discharge curve has a fast kinetic plateau at approximately 1.4 V and a slow kinetic plateau at approximately 1.3 V, which correspond to the generation of MnOOH upon H+ insertion and the formation of ZnMn2O4 upon Zn2+ insertion, respectively [Figure 1C]. Without Zn2+ electrolyte, only the H+ insertion plateau is retained. The non-aqueous electrolyte has almost no capacity due to the lack of H+, and its capacity recovers after adding water. The ex-situ XRD shows that the characteristic peak of MnOOH appears at 1.3 V when the discharge is stopped, and the characteristic peak of ZnMn2O4 appears at 1.0 V when the discharge continues further [Figure 1D]. Combined with the elemental mapping results, it further proves that the energy storage mechanism of this Zn/MnO2 battery is the continuous reversible intercalation/deintercalation of H+ and Zn2+ [Figure 1E].
Zhao et al.[70] reported a synergistic H+/Zn2+ insertion mechanism in battery systems. The novel manganese oxide nanosheets MON [chemical formula MnO2H0.16(H2O)0.27] achieve stable ion insertion/extraction through their monoclinic P21/c(14) space group and 1 × 1 tunnel structure.
Chemical conversion reaction mechanism
The aforementioned mechanisms all center on Zn2+ insertion, whereas Pan et al. proposed a novel concept emphasizing that the reversible chemical conversion between α-MnO2 and H+ is the key process[71]. During discharge, metallic zinc undergoes oxidation at the negative electrode, releasing electrons and dissolving into the electrolyte as Zn2+ ions. This process is fully reversible during charging: MnOOH is reoxidized to α-MnO2 at the positive electrode, while Zn2+ ions are simultaneously reduced and electrodeposited as metallic zinc at the negative electrode[71]. They directly observed through transmission electron microscopy (TEM) that during the discharge process, α-MnO2 transformed from nanofibers into short nanorods and nanoparticle aggregates with characteristic interplanar spacings of MnOOH [Figure 2A]. After charging, the structure could be restored to α-MnO2, proving that the positive electrode undergoes reversible structural evolution and transformation reactions. Combined with scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (STEM-EDS) element mapping, it was found that the discharge products only contained Mn and O, and no Zn element. Zn was only distributed in the lamellar by-products, directly confirming that Zn2+ did not embed into the MnO2 lattice [Figure 2B]. Additionally, XRD characterization was used to confirm the appearance of characteristic diffraction peaks of MnOOH and ZnSO4[Zn(OH)2]3·xH2O in the discharged state, without the characteristic peaks of zinc ion insertion phase. Multiple characterizations jointly confirmed that the energy storage mechanism of this aqueous Zn/MnO2 battery is the reversible transformation reaction between MnO2 and H+ (MnO2 ↔ MnOOH), rather than the traditional understanding of the reversible insertion/deletion mechanism of Zn2+.

Figure 2. (A) MnO2 electrodes discharged to 1 V. The arrow indicates the growth directions of the short nanorods; (B) STEM-HAADF image of short nanorods and STEM-EDS mappings of the elemental distributions of Mn, O and Zn in the MnO2 electrode in the discharged state during the first cycle. (A and B) Reprinted with permission from[71]. Copyright 2016, Springer Nature. STEM-HAADF: Scanning transmission electron microscopy-high-angle annular dark-field; STEM-EDS: scanning transmission electron microscopy-energy dispersive X-ray spectroscopy.
Furthermore, Liu et al.[72] found that during the discharge process of ZIBs, β-MnO2 first reacts with protons in the electrolyte to form MnOOH which further reacts with protons to form dissolved Mn2+. In the subsequent cycle, the reaction stabilizes as the mutual transformation between ε-MnO2 and MnOOH or
Deposition-dissolution mechanism
In zinc-ion batteries, the deposition-dissolution mechanism refers specifically to the reversible electrochemical processes at the metallic zinc anode, involving the reduction deposition (electroplating) and oxidation dissolution (stripping) of Zn2+ at the electrode/electrolyte interface during charge-discharge cycles[73]. The MnO2 cathode achieves a high theoretical specific capacity of 616 mAh g-1 through a redox reaction involving double electron transfer[74]. It is worth noting that this reaction mechanism was proposed as early as the late 20th century[75]. Recently, Wu et al.[76] employed in situ Raman microscopy and other techniques to investigate the previously unclear relationship between Mn3+ Jahn-Teller distortion and Mn-O bond length changes, as well as the distortion caused by post-processing in ex-situ characterization. They achieved the first in-situ observation of manganese Jahn-Teller distortion during operation, while also directly revealing the mechanism of dissolution-redeposition core reaction mechanism of MnO2 cathodes. Furthermore, it was demonstrated that the addition of MnSO4 to the electrolyte effectively suppresses manganese dissolution and significantly improves the cycling stability of the battery.
In 2022, Chen et al.[77] investigated the long-standing controversy surrounding the energy storage mechanism of aqueous zinc-manganese (Zn-Mn) batteries. They challenged the conventional view that zinc sulfate hydroxide [Zn4SO4·(OH)6·xH2O, ZSH] as merely a byproduct. They proposed a ZSH-assisted deposition-dissolution reaction model for aqueous Zn-Mn batteries in mildly acidic sulfate electrolytes. The system elucidates the ZSH-assisted deposition-dissolution reaction mechanism: In a mildly acidic sulfate electrolyte containing Mn2+, charging to ~1.5 V induces a two-electron transfer reaction between ZSH and Mn2+, forming low-crystallinity ZnxMnO(OH)2 nanosheets[77]. During discharge, ZnxMnO(OH)2 nanosheets dissolve via proton diffusion reactions, while localized pH changes in the electrolyte trigger ZSH regeneration, completing a reversible cycle. This reaction constitutes the core mechanism for aqueous Zn-Mn battery energy storage. To validate this model, a MnO2-free Zn-ZSH battery was constructed, exhibiting identical electrochemical behavior and phase evolution to the original battery[77]. These findings directly corroborate the pivotal role of ZSH.
Key materials for Zn-ion batteries
Cathode material for Zn-ion batteries
In the technological development of ZIBs, cathode materials serve as a pivotal determinant for regulating electrochemical energy storage, as their intrinsic properties directly dictate the device’s energy density and operational efficiency. To date, extensively investigated cathode materials for ZIBs can be broadly categorized into manganese Mn-based materials, vanadium V-based materials, Prussian blue analogues (PBAs), among others. The following sections will systematically review these major material classes alongside their respective modification strategies.
Manganese-based compounds have attracted substantial research interest owing to their abundance, exceptional environmental compatibility, high cost-effectiveness, low toxicity, and rich valence states. Among various candidates, MnO2 stands out as a premier choice; beyond its economic and ecological merits, it delivers superior electrochemical performance, characterized by a high theoretical specific capacity and an elevated discharge voltage plateau. Furthermore, it exhibits exceptional compatibility with aqueous electrolytes and possesses an optimal redox potential window[78]. α-MnO2, β-MnO2, and γ-MnO2 all possess tunnel structures, thus allowing Zn2+ to reversibly insert and extract[79-82].
α-MnO2 possesses a one-dimensional (1D) 2 × 2 tunnel-like micro-channeled framework that facilitates Zn2+ deintercalation, thereby enabling rapid ion diffusion. Recently, Li et al.[80] addressed the challenges of kinetic retardation and poor cycling stability associated with α-MnO2 cathodes in ZIBs. They designed a magnesium-doped α-MnO2 (MMO) structure featuring a low concentration of oxygen vacancies. This strategy effectively suppressed manganese dissolution; consequently, the MMO cathode demonstrated significantly superior performance compared to bare α-MnO2 after 700 cycles [Figure 3A]. Furthermore, an evaluation of Mn dissolution revealed that after 100 cycles, the Mn/Zn mass ratio for the MMO-2 electrode was restricted to 0.086, whereas it reached 0.137 for bare α-MnO2 [Figure 3B]. Tian et al.[83] developed a PVP-Al-MnO2 cathode by co-intercalating Al3+ and polyvinylpyrrolidone (PVP) into the tunnel structure of α-MnO2 The resulting material delivered a specific capacity of 306.8 mAh g-1 at 0.3 A g-1, alongside an impressive capacity retention of 93.1% over 2,000 cycles [Figure 3C]. Han et al.[84] incorporated Eu into β-MnO2 (20EM) as the cathode material for ZIBs. Eu doping increases the lattice spacing and enhances the oxygen vacancy content. The prepared 20EM achieved a capacity of 409 mA h g-1 after 42 cycles at 0.2 A g-1 [Figure 3D]. After 1,000 cycles, its performance remains stable [Figure 3E].
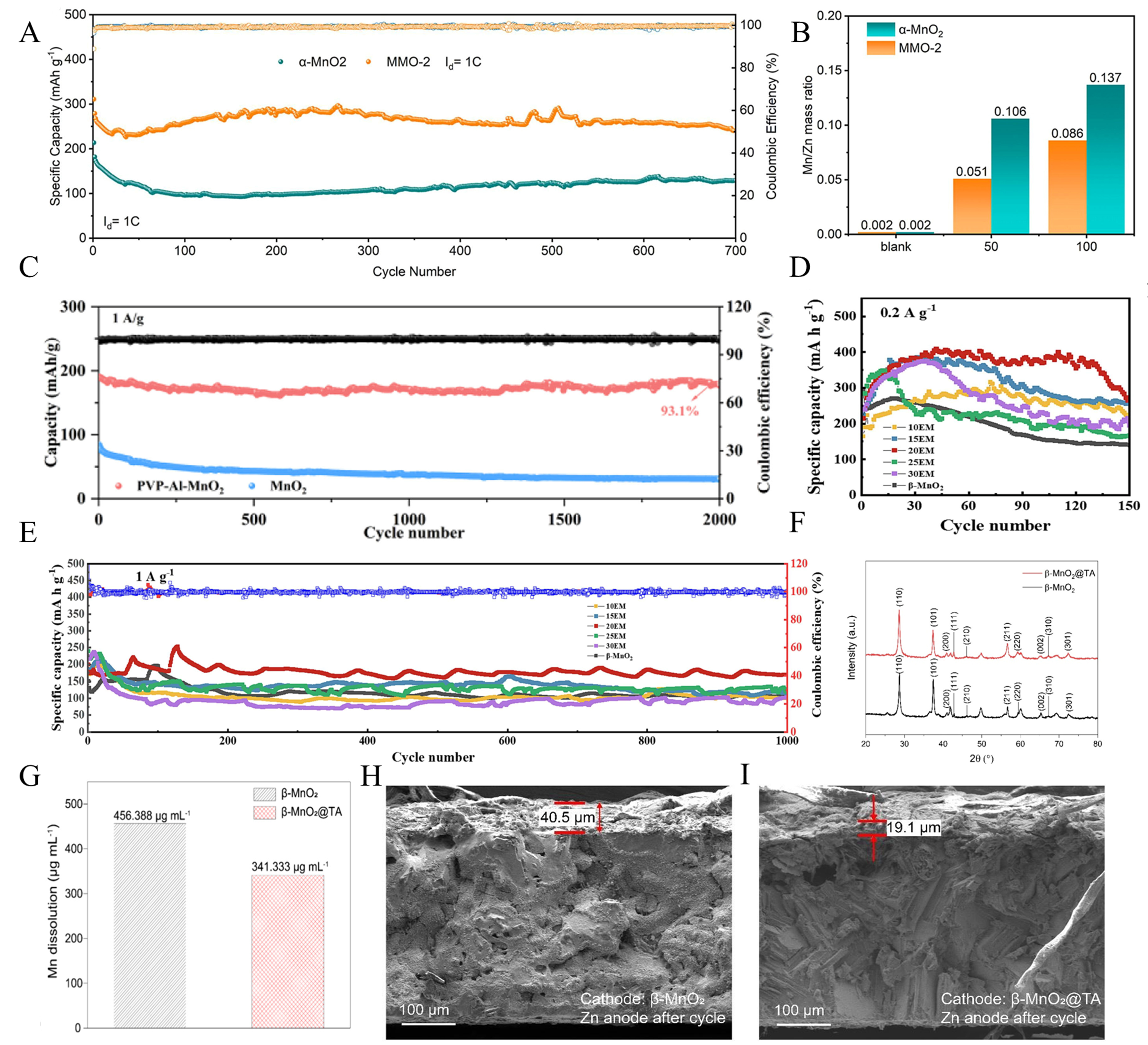
Figure 3. (A) Cycle Performance Comparison of Bare MnO2 and MMO-2 at 1C Rate. (B) Mn/Zn ratio in electrolyte for both bare MnO2 and MMO electrodes at different cycles. (A and B) Reprinted with permission from[80]. Copyright 2024, Elsevier; (C) Cycle performance of PVP-Al-MnO2 at 1.0 A g-1. Reprinted with permission from[83]. Copyright 2024, Elsevier; (D) Rate performance at different current densities. (E) Cycling performance at 1 A g-1 of β-MnO2 and Eu-doped MnO2 with different molar concentrations. (D and E) Reprinted with permission from[84]. Copyright 2023, Elsevier; (F) XRD patterns of synthesized β-MnO2 and β-MnO2@TA powder. (G) ICP analysis results comparing β-MnO2 and β-MnO2@TA after 100 cycles. (H-I) Cross-section SEM images of (H) Zn metal anode after 100 cycles using β-MnO2 as a cathode, (I) Zn metal anode after 100 cycles using β-MnO2@TA as a cathode. (F-I) Reprinted with permission from[85]. Copyright 2024, American Chemical Society. MMO: Mg-doped α-MnO2 configuration; XRD: X‑ray diffraction; TA: tannic acid; SEM: scanning electron microscopy.
Key issues in ZIBs involving Mn2+ leaching, phase transformations, and zinc dendrite formation in β-MnO2 cathodes. Paik et al.[85] proposed the use of tannic acid (TA), a hydroxyl-rich polyphenolic biomolecule, as a protective coating for β-MnO2. Powder X-ray diffraction (PXRD) analysis confirmed that the TA coating preserved the intrinsic crystal structure of β-MnO2 [Figure 3F]. Furthermore, the TA layer effectively mitigated manganese dissolution into the electrolyte [Figure 3G] and accelerated the phase transition kinetics during charge-discharge cycling. Notably, employing β-MnO2@TA as the cathode also led to the successful suppression of zinc dendrite formation over 100 cycles [Figure 3H-I].
The γ-MnO2 is classified as one of the orthorhombic crystal system, exhibiting low crystallinity with numerous defects within its lattice structure. From a microstructural perspective, the tunneling structure of this material is not regular in form. Instead, it is jointly composed of 1 × 1 tunnels characteristic of manganite and 1 × 2 tunnels typical of rhodochrosite, arranged in an irregular alternating pattern[86]. Despite this considerable structural complexity, γ-MnO2 delivers exceptional energy storage performance. Du et al.[87] synthesized a composite material (NCM) comprising amino-functionalized multi-walled carbon nanotubes (MWCNTs-NH2) and γ-MnO2 via a hydrothermal method. The introduced amino groups not only interact with H+/Zn2+ to provide additional pseudocapacitance but also promote the redeposition of Mn2+ through strong adsorption, thereby compensating for the loss of active material. Consequently, the NCM electrode delivered outstanding electrochemical performance. After 120 cycles, it achieved a high specific capacity of 417.9 mAh g-1 with a capacity retention exceeding 100% [Figure 4A]. Furthermore, the NCM demonstrated remarkable long-term cycling durability, maintaining a capacity retention of 91.4% over 3,500 cycles [Figure 4B]. Additionally, Gan et al.[88] synthesized high-valent tungsten (W6+)-doped MnO2 nanosheets (W-MnO2). By utilizing W-doping to modulate the MnO2 crystal structure, they facilitated its transformation from the β to the γ phase. Simultaneously suppressing the dissolution of Mn3+. At a doping ratio of 1:0.01, the redox reaction of W-MnO2 is more complete[88]. The discharge capacity at 0.1 A g-1 reaches 240.6 mAh g-1. In comparison with W-MnO2 samples with different doping ratios at an equivalent current density, the 1:0.01 sample exhibited superior cycle life and augmented discharge specific capacity [Figure 4C]. After 1,000 cycles at 1 A g-1, the capacity retention reached 79.8% [Figure 4D].
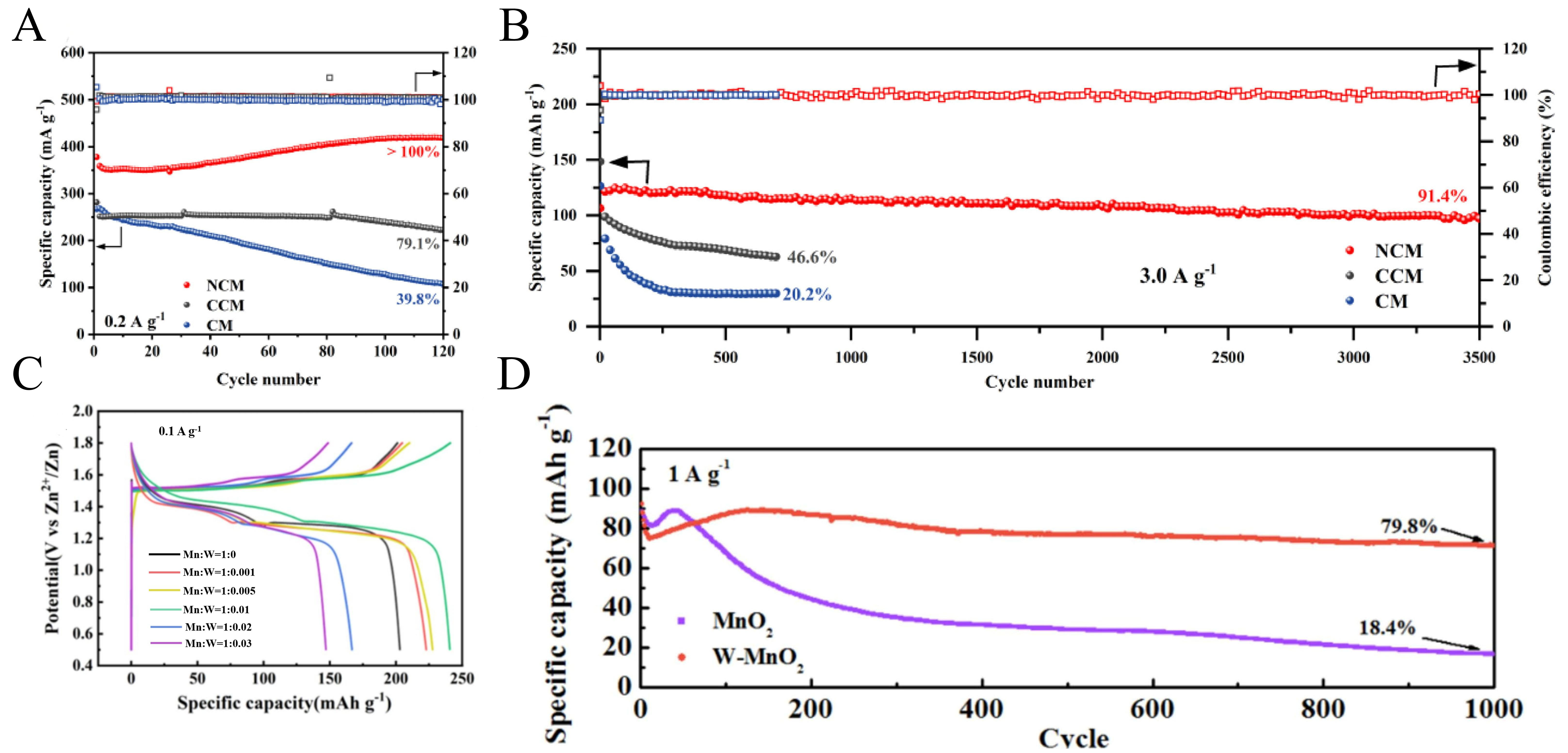
Figure 4. (A) Cycling performance of NCM at 0.2 A g-1. (B) Cycling performance of NCM at 3.0 A g-1. (A and B) Reprinted with permission from[87]. Copyright 2023, Elsevier; (C) GCD curves of MnO2 with different W doping ratio at 0.1 A g-1. (D) Long-cycle performance of MnO2 and W-MnO2. (C and D) Reprinted with permission from[88]. Copyright 2025, Elsevier. NCM: MWCNTs-NH2/γ-MnO2 nanocomposite; CCM: MWCNTs-COOH/MnO2 composites; CM: MWCNTs/MnO2 composites; GCD: galvanostatic charge-discharge.
In 2024, Paudel et al.[89] investigated MnO2 cathodes for rechargeable alkaline Zn-MnO2 batteries. Although nanostructuring and the introduction of crystal defects enhance electrochemical performance, the underlying atomic-scale mechanisms remain elusive. Previous studies have predominantly focused on bulk MnO2, lacking a systematic comparative analysis across its mainstream crystallographic polymorphs. To address this gap, we employed first-principles density functional theory (DFT) calculations to construct bulk models of β-MnO2 (pyrolusite), R-MnO2 (ramsdellite), and γ-MnO2 featuring manganese and oxygen vacancies, alongside low-energy surface models for these three polymorphs. By simulating the core processes of MnO2 discharge in alkaline batteries, key parameters such as defect formation energy, H+ binding energy, and unit cell volume changes were calculated and analyzed. Results indicate that Mn and O vacancy formation energies are higher in β-MnO2, while R-MnO2 and γ-MnO2 exhibit greater defect formation propensity due to the presence of 2 × 1 large tunnels[89]. Specifically, Mn vacancies can delay H+ filling of the
Vanadium (V)-based materials exhibit remarkable crystallographic diversity, with distinct structural features conferring unique advantages in electrochemical performance. Among these, layered structures significantly minimize diffusion resistance during ion migration via their open two-dimensional pathways, establishing the structural foundation for fast charge/discharge kinetics. Conversely, tunnel structures - owing to their highly rigid and stable frameworks - effectively mitigate volumetric strain during cycling, thereby ensuring exceptional cycling stability. From a mechanistic perspective, the multivalent nature of vanadium facilitates multi-electron redox reactions, which serves as the fundamental origin of their extraordinarily high specific capacities.
Tunnel-type vanadium oxides have emerged as promising cathode materials for ZIBs. Nevertheless, in contrast to their layered counterparts which benefit from tunable interlayer spacings, accelerating ion transport kinetics within these rigid, fixed-size tunnels remains a formidable challenge. To address this limitation, He et al.[90] synthesized two distinct VO2 nanostructures by tailoring the ratio of V2O5 to glucose precursors. They systematically investigated the influence of macroscopic architectural arrangement within the electrode on tunnel orientation and subsequent ion transport. Their findings revealed that VO2-D nanoribbons preferentially aligned parallel to the electrode surface and stacked along the c-axis, thereby constructing highly directional ion-transport channels. In contrast, the VO2-A counterpart exhibited a randomly oriented configuration, resulting in isotropic ion transport. The distinct advantage of this macroscopic architectural design lies in its ability to overcome the sluggish ion diffusion inherent to tunnel-type vanadium oxides, without necessitating any modifications to the intrinsic crystal structure. Furthermore, the relatively low specific surface area of the VO2-D nanoribbons (17.50 m2 g-1) minimizes detrimental interfacial contact with the electrolyte. This restricted interfacial exposure effectively mitigates vanadium dissolution, thereby significantly enhancing the overall cycling stability[90]. Additionally, VO2-D exhibits excellent performance, achieving a capacity of 420.8 mAh g-1 at 0.1 A g-1 and maintaining
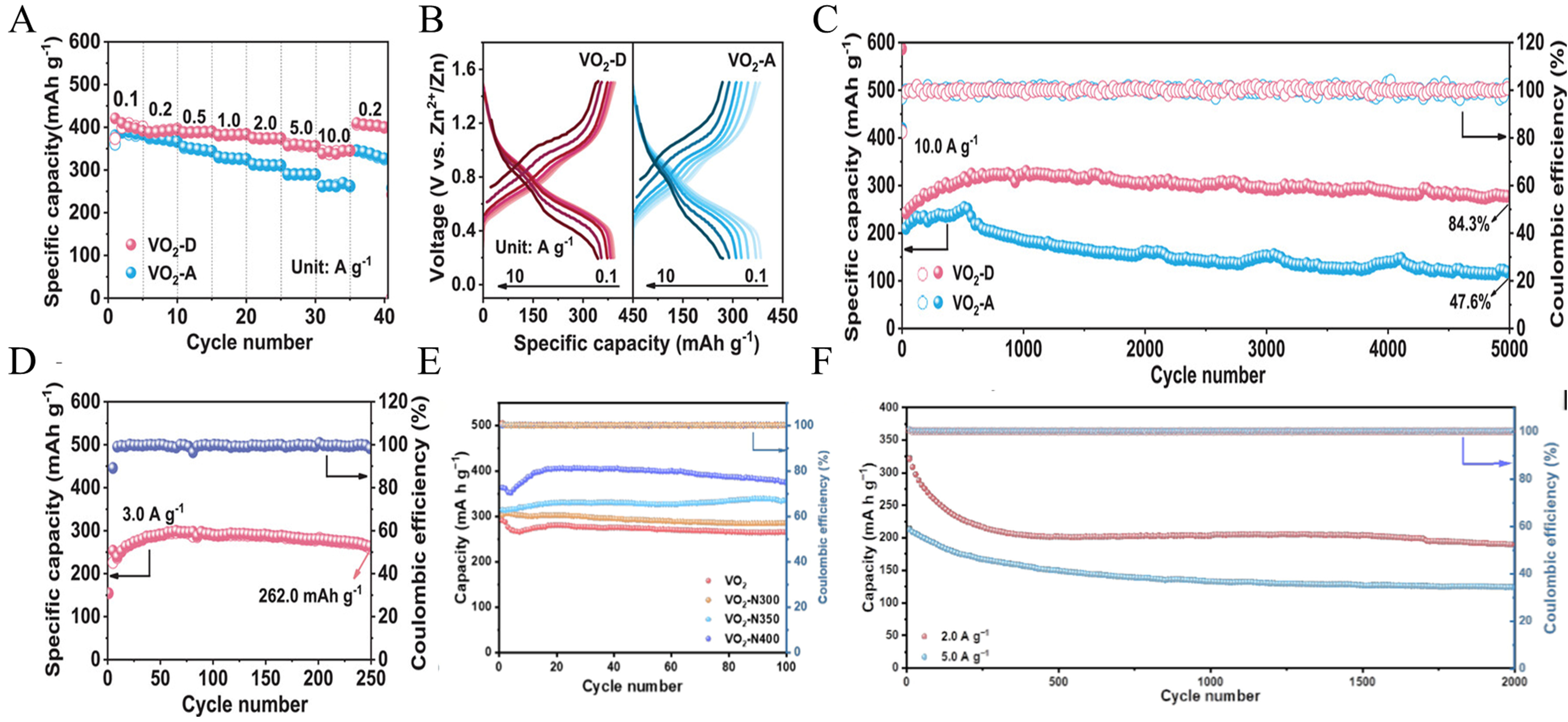
Figure 5. (A) Rate Performance at Current Densities Ranging from 0.1 A g-1 to 10 A g-1. (B) Galvanostatic charge/discharge profiles at the current densities from 0.1 A g-1 to 10 A g-1. (C) Cycling performance for VO2-D and VO2-A cathodes at 10 A g-1. (D) Cycling performance of Zn//VO2-D soft-packaged battery at 3 A g-1. (A-D) Reprinted with permission from[90]. Copyright 2024, Wiley; (E) Cycling performances and corresponding CEs of VO2-N400 at the current density of 0.1 A g-1. (F) Long cycling performances of VO2-N400 at 2 and 5 A g-1. (E and F) Reprinted with permission from[54]. Copyright 2023, Elsevier. CEs: Coulombic efficiencies.
The inherently low electronic conductivity of vanadium-based compounds, coupled with the sluggish diffusion kinetics of Zn2+ within their crystal lattices, collectively constitute the primary bottlenecks impeding their transition toward practical applications. Gu et al.[54] synthesized nitrogen-doped VO2(B) nanoribbons (VO2-N) via an ammonia thermal treatment strategy. The introduction of nitrogen dopants induces lattice expansion and grain refinement within the VO2(B) matrix, generating a disordered structure that provides abundant ion storage sites and continuous transport pathways. Consequently, this approach narrows the bandgap of VO2 from 1.3 eV to 0.7 eV, thereby boosting intrinsic electronic conductivity, while simultaneously drastically lowering the Zn2+ diffusion energy barrier from 2.86 eV to 0.29 eV. As a result, the optimized VO2-N400 electrode exhibits exceptional electrochemical performance, delivering a specific capacity of 373.7 mAh g-1 after 100 cycles without any capacity degradation [Figure 5E]. Furthermore, the assembled battery demonstrated robust long-term cycling stability, maintaining discharge capacities of
Vanadium pentoxide (V2O5) is a well-known cathode material and serves as a typical example of layered V-based oxides. Li et al.[91] employed an interfacial coupling strategy to synthesize porous carbon. This material was then composite with V2O5 to construct a cathode material for aqueous Zn-V batteries. The interface coupling between layered V2O5 and porous carbon forms a 3D conductive network. The porous carbon enhances interfacial conductivity and shortens ion insertion-deintercalation pathways, while V2O5 provides Zn2+ transport pathways and active sites. This composite material exhibits outstanding performance, achieving a specific capacity of 244.4 mAh g-1 and an energy density of 171 Wh kg-1 at 50 mA g-1 [Figure 6A]. After 7,000 cycles, the capacity retention reached 92% with a CE of nearly 98% [Figure 6B]. Wen et al.[92] achieved dual structural and electronic control of V2O5 through H2SO4 etching combined with hydrothermal annealing, preparing V2O5-H2SO4 cathode materials with an interlaced nanowire structure. Concurrently, a protective layer comprising CuZn2/In/InZn3 was deposited on the zinc foil surface, thereby impeding the growth of zinc dendrites. The etching strategy significantly increases the specific surface area of V2O5-H2SO4 compared to pristine V2O5. The anode protective layer further reduces battery polarization and charge transfer resistance. Meanwhile, the V2O5-H2SO4 half-cell achieved a specific capacity of 515.2 mAh g-1 at
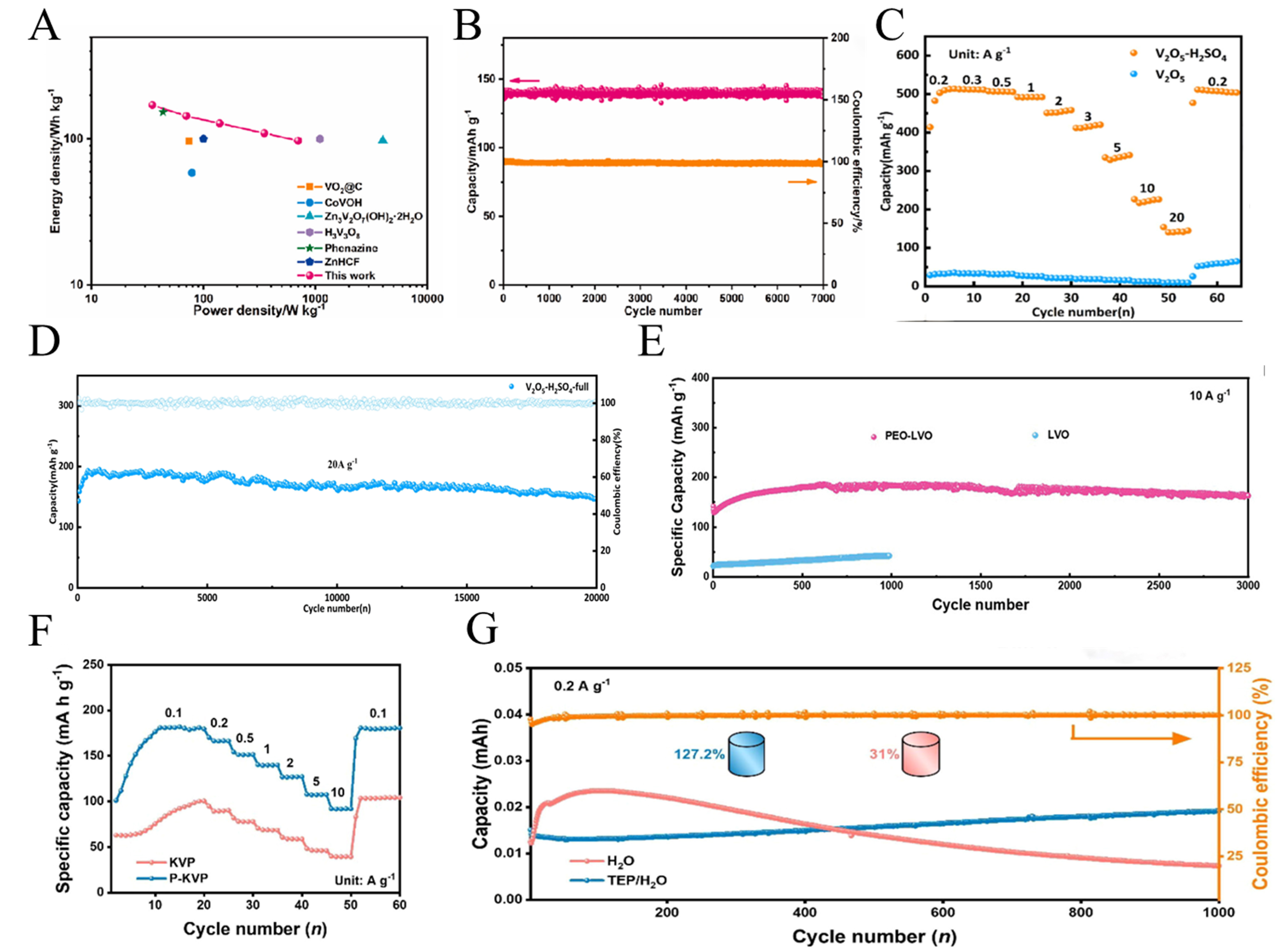
Figure 6. (A) Ragone plots of the V2O5-180. (B) Stability and Coulombic efficiency of V2O5-180 for 7,000 cycles at 1.0 A g-1. (A and B) Reprinted with permission from[91]. Copyright 2024, Elsevier; (C) Rate performance at 0.2 A g-1 with coulombic efficiency of V2O5 and V2O5-H2SO4. (D) Cycling performance of full-battery at 0.2 A g-1. (C and D) Reprinted with permission from[92]. Copyright 2025, Elsevier; (E) Cycling durability of LVO and PEO-LVO cathodes at 10 A g-1. Reprinted with permission from[93]. Copyright 2024, Wiley; (F) Rate performance of KVP and P-KVP electrode under different current densities ranging from 0.1 to 10 A g-1. (G) Long-term cycling tests and coulombic efficiency of P-KVP and KVP cathodes in different electrolytes at 0.2 A g-1. (F and G) Reprinted with permission from[94]. Copyright 2024, Elsevier. LVO: Pure LiV3O8; PEO-LVO: poly(ethylene oxide)-LiV3O8 superlattice nanosheets; KVP: pristine K0.5VOPO4·1.5H2O; P-KVP: porous K0.5VOPO4·1.5H2O polyanionic cathode.
Wu et al.[93] prepared PEO-LiV3O8 (PEO-LVO) superlattice nanosheet cathodes via a simplified hydrothermal method. By inserting PEO molecules into the interlayer space of LiV3O8 (LVO), forming an alternating “LVO monolayer-PEO chain layer” stacking structure[93]. PEO reduces the diffusion barrier for Zn2+, increases active sites, and enables highly reversible H+/Zn2+ co-intercalation. This reduces alkaline byproducts on the cathode surface and accelerates interfacial kinetics. At 10 A g-1, the PEO-LVO anode still demonstrates a reversible capacity of 163.1 mAh g-1, maintaining an excellent performance after 3,000 cycles [Figure 6E]. Wang et al.[94] prepared porous K0.5VOPO4·1.5H2O (P-KVP) polycationic cathode. SXRD and XPS confirmed that energy storage occurs through sequential solid solution intercalation and phase transformation during Zn2+ storage, accompanied by anisotropic crystal plane expansion[94]. To address the voltage drop caused by irreversible conversion to amorphous VOx in the P-KVP cycle, a 2 M Zn(OTF)2/triethyl phosphate (TEP)-water (4:1) mixed electrolyte was introduced. This inhibits the conversion by forming a phosphorus-rich electrolyte interface layer. P-KVP exhibits specific capacities ranging from 181.8 mAh g-1 to 92.0 mAh g-1 at current densities of 0.1-10 A g-1 [Figure 6F]. After 2,000 cycles, the capacity retention rate reached 94.2% with a CE close to 100%. After optimization, stable high-voltage output exceeding 1.2 V was maintained even after 1,000 cycles [Figure 6G]. Flexible fiber-shaped ZIBs fabricated from this material exhibited stable performance under bending angles ranging from 0° to 180°.
Within the expanding landscape of cathode materials for ZIBs, PBAs have demonstrated outstanding potential alongside traditional Mn- and V-based compounds, emerging as a prominent research frontier. Hexacyanometalates, the fundamental building blocks of PBAs, feature a robust three-dimensional (3D) open-framework structure. Benefiting from the spatial accommodation provided by this architecture for Zn2+ migration, zinc ions can undergo highly reversible intercalation and deintercalation processes within the crystal lattice. These unique structural advantages have firmly established PBAs as a focal point in the ongoing development of advanced ZIB cathodes. The chemical formula of PBAs is AxM[M’(CN)6]1-yγy·wH2O, where A represents an alkali metal ion, M and M’ are transition metal elements, and γ is the hexacyanometallate complex vacancy[95].
Despite the immense promise of PBA materials, their practical electrochemical performance in ZIBs often exhibits substantial variability. Syed et al.[96] synthesized copper-substituted manganese Prussian blue analog (CuMn PBA) composite nanostructures with varying copper (Cu) and manganese (Mn) concentrations. CuMn PBA-2 exhibits a monoclinic crystal structure, high crystallinity, and uniform distribution of elements such as Cu, Mn, and Fe. Its larger specific surface area and suitable loose structure enhance contact between the electrolyte and active material. Partial substitution of Cu and Mn vacancy coordination suppress Jahn-Teller distortion to stabilize the structure. CuMn PBA-2 performed exceptionally well, it maintained excellent rate capability of 89.08 mAh g-1 at 2.4 A g-1[96]. After 2,000 cycles at 3 A g-1, the capacity remained at 73.15 mAh g-1 with a coulombic efficiency close to 100% [Figure 7A]. Yang et al.[97] proposed constructing an epitaxial core-shell Mn@FeHCF cathode via acid etching-ion exchange, achieving epitaxial growth through lattice matching. The FeHCF shell layer provides low-strain support to suppress Jahn-Teller distortion in the MnHCF core layer, while the surface amorphous layer acts as a protective barrier. This approach avoids the capacity and voltage trade-offs associated with conventional strategies. Its performance is outstanding, with a capacity retention rate of 72.4% after 400 cycles, and a discharge capacity of 117 mAh g-1 at 2 A g-1 with an 83.9% capacity retention after 4,800 cycles [Figure 7B]. Furthermore, the 11.86 mAh pouch cell operates stably within -30 °C to 60 °C [Figure 7C].
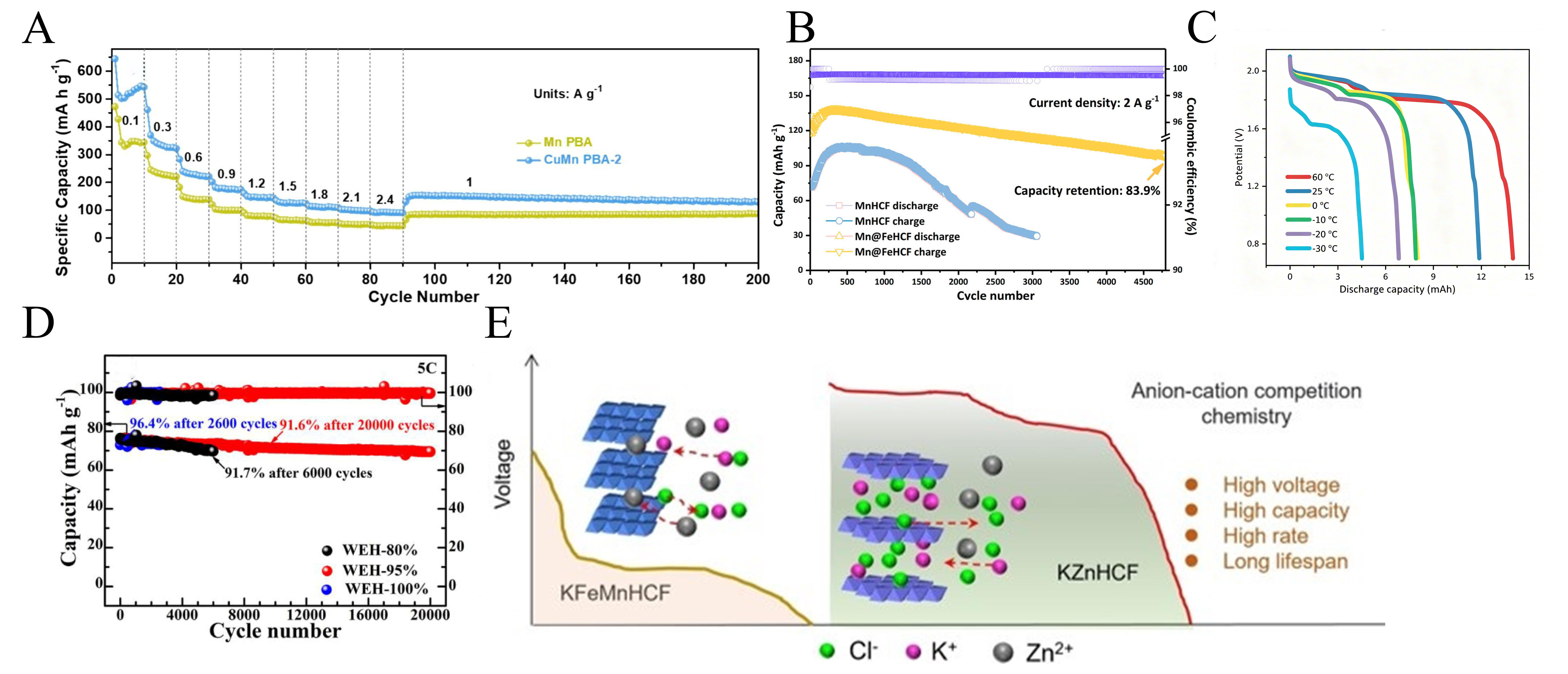
Figure 7. (A) Rate performance comparison of Mn PBA and CuMn PBA-2 electrodes at various current densities. Reprinted with permission from[96]. Copyright 2024, Elsevier; (B) Cycling performance at 2 A g-1. (C) Discharge voltage profiles and cycling performance of Zn||Mn@FeHCF cell at 0.03 A g-1 under different circumstances. (B and C) Reprinted with permission from[97]. Copyright 2023, American Chemical Society; (D) long-term cycling stability of cells at 5C with WEH-80%, WEH-95% and WEH-100%. Reprinted with permission from[98]. Copyright 2022, Elsevier. (E) Illustration of the anion-cation competition chemistry. Reprinted with permission from[52]. Copyright 2024, Wiley. PBA: Prussian blue analogue.
Sun et al.[98] addressed the practical limitations of rechargeable ZIBs due to zinc corrosion, zinc dendrite growth, and cathode dissolution. They proposed a Daniell-type zinc/Prussian blue (MnPB) battery featuring a MnPB cathode and zinc anode, employing a water-ethanol hybrid (WEH) electrolyte at standard salt concentrations to overcome these challenges. Containing 95 wt% The WEH electrolyte containing 95 wt% ethanol enables the battery to exhibit outstanding performance, achieving a capacity retention of 91.6% after 20,000 cycles at 5C [Figure 7D]. Cui et al.[52] addressed the issues of low discharge capacity and poor rate performance in PBA cathodes caused by the traditional reliance on cation (de)intercalation mechanisms. They firstly proposed and systematically investigated high-voltage scan-triggered anion-cation competition chemistry. Using KFeMnHCF synthesized by the coprecipitation method as the cathode, the battery achieves comprehensive superior performance in a dilute KCl + ZnSO4 electrolyte through this competitive mechanism [Figure 7E]. At 0.6 A g-1 to 5 A g-1, the capacity retention exceeds 96%, with cycle stability surpassing 3,000 cycles and a CE of approximately 80%. Following the phase transition, the electronic conductivity of KZnHCF increases, the bandgap decreases from 1.15 eV to 0.99 eV, and the reduction in diffusion energy barriers for Cl- and K+ is key to the performance enhancement.
In aqueous ZIBs, the cathode-electrolyte interphase (CEI) plays a critical role in dictating the structural integrity of cathode materials and the long-term cycling performance of the battery. The formation and evolution of the CEI are intricately linked to the oxidative decomposition of the electrolyte and the surface reconstruction of the active material during cycling. For Mn-based cathodes, the Jahn-Teller distortion of Mn3+ during charge/discharge processes induces irreversible phase transitions (e.g., from layered δ-MnO2 to spinel ZnMn2O4) and triggers severe Mn2+ dissolution. Furthermore, these structural degradation mechanisms are concurrently accompanied by the continuous oxidation of water and electrolyte anions at the cathode surface[99,100]. The accumulation of amorphous MnOx, sulfate precipitates, and other parasitic byproducts results in the formation of a heterogeneous and unstable CEI on the cathode surface. This defective interphase fails to effectively suppress Mn2+ dissolution; worse still, it obstructs the Zn2+ transport pathways, ultimately leading to rapid capacity degradation[101]. Similarly, for V-based cathode materials, the continuous dissolution of active vanadium species into the aqueous electrolyte, coupled with the accumulation of electrochemically inert byproducts, induces the formation of a defective CEI. This compromised interfacial layer constitutes the primary root cause of capacity fading in V-based systems[102].
The surface reconstruction of ZIBs cathodes acts as a “double-edged sword” with respect to electrochemical performance. On the one hand, irreversible reconstructions - such as deleterious phase transitions in MnO2 and the amorphization of vanadium-based materials - can lead to severe structural collapse and the loss of active materials. On the other hand, controlled in situ surface reconstruction can facilitate the formation of a robust and ion-conductive CEI. For instance, a tannic acid coating on β-MnO2 induces the generation of a uniform CEI enriched with Mn-O-C bonds; this not only suppresses Mn2+ dissolution but also accelerates phase transition kinetics during cycling[85]. Furthermore, electrolyte additives, such as MnSO4 and sodium carboxymethyl cellulose, can regulate CEI formation via specific adsorption onto the cathode surface, thereby effectively inhibiting the oxidative decomposition of water and the dissolution of transition metal ions[103,104].
Anode material for Zn-ion batteries
The inherent instability of zinc anodes primarily stems from inhomogeneous Zn2+ plating/stripping behaviors and parasitic side reactions during charge-discharge cycling, which collectively manifest as three major interconnected issues. Specifically, dendrite growth originates from the non-uniform deposition of Zn2+ onto the anode surface. Fundamentally, this process is driven by localized fluctuations in the electric field distribution and Zn2+ concentration gradients at the electrode interface. According to the Chazalviel-Brissot space charge theory, once the applied current density exceeds a critical threshold (i.e., the limiting current density), the Zn2+ concentration at the anode-electrolyte interface rapidly depletes to zero[105,106]. The resulting localized space-charge accumulation generates a substantial electric field, driving the preferential deposition of Zn at protruding tips and initiating dendrite growth. Furthermore, continuous dendrite propagation can ultimately pierce the separator, triggering catastrophic internal short circuits within the battery. Simultaneously, this highly porous morphology significantly amplifies the electrochemically active surface area of the anode, thereby accelerating parasitic side reactions. Additionally, the intrinsic microscopic roughness of the zinc electrode and the continuous rupture of the brittle solid electrolyte interphase (SEI) layer synergistically exacerbate this vicious cycle of dendrite proliferation.
To address zinc dendrites and side reactions in zinc-based batteries, researchers have made numerous efforts. Hu et al.[107] constructed a highly crystalline, flower-like two-dimensional acetylene-based covalent organic framework (COF-H) as an artificial interfacial layer on zinc anodes, synergistically inhibiting dendrite growth and corrosion through both structural design and mechanism of action. It physically isolates the Zn anode from contact with the electrolyte, preventing self-corrosion induced by water/oxygen-induced self-corrosion and HER. Linear polarization testing reveals its corrosion current (20.0 mA cm-2) is apparently lower than that of a bare zinc electrode, with the surface remaining intact after 10 days of immersion[107]. Furthermore, the acetylene group, ketone group, and enamine group in COF-H exhibit strong affinity for Zn2+. The nucleation overpotential of the COF@Cu electrode is only 23 mV [Figure 8A]. Simultaneously, the ordered micropores guide uniform transport and deposition of Zn2+, preventing dendrite nucleation caused by localized overconcentration. Scanning electron microscope (SEM) observations reveal that the surface of the cycled COF@Zn electrode exhibits dense, planar zinc layers, whereas the bare zinc electrode is covered with vertical dendrites [Figure 8B]. The battery exhibited a cycle life exceeding 900 hours at 3 mA cm-2, with retention rate of 73.7% after 300 cycles [Figure 8C]. Zong et al.[108] employed diethyl phosphoramidate (DP) as a multifunctional electrolyte additive to optimize cathode interfacial chemistry, modulate the electrolyte, and achieve formation an organic-inorganic hybrid solid electrolyte interphase at the anode [Figure 8D]. Its molecules can break the hydrogen bond of H2O and adsorb onto the zinc surface, inducing the formation of a robust crystalline-amorphous hybrid SEI layer composed of ZnS, Zn3N2, and Zn3(PO4)2. At the cathode surface, a DP-rich dynamic CEI forms, preventing the dissolution of V-based materials caused by H2O while enhancing intercalation stability and reversibility. Therefore, the Zn||Zn symmetric cell achieved 1,200 h at 10 mA cm-2 and a surface capacity of 5 mAh cm-2 [Figure 8E]. The Zn||Cu asymmetric battery maintained CE of 99.7% after 800 cycles at 1 mA cm-2 and 0.5 mAh cm-2 [Figure 8F]. Furthermore, the Zn||NVO full cell exhibited a capacity retention rate of 87% after 1,000 cycles at 4 A g-1 [Figure 8G], and the assembled pouch cell maintained stable operation beyond 200 cycles.
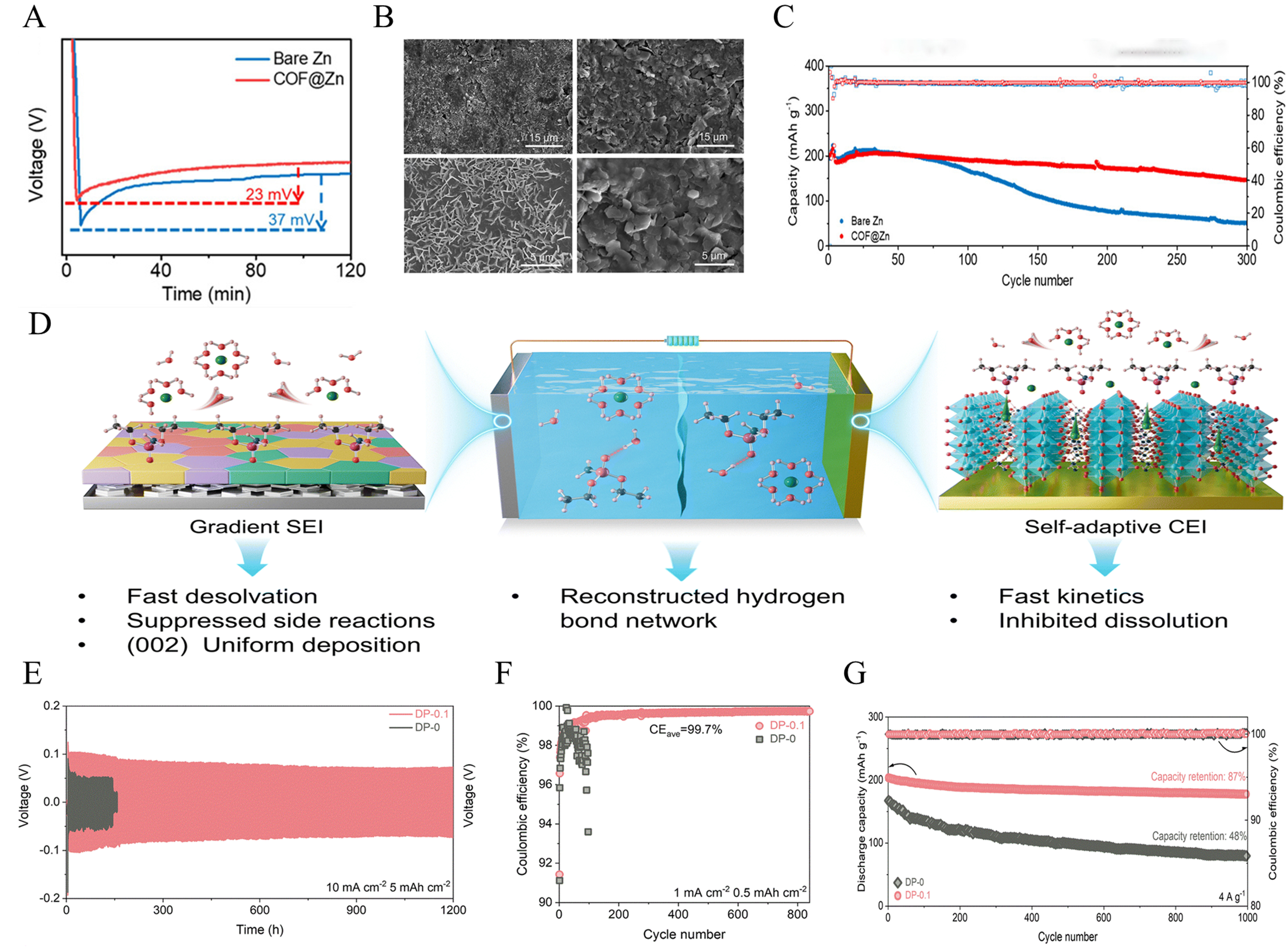
Figure 8. (A) Nucleation Overpotential Comparison of Zn Deposition on COF@Cu and Bare Cu Electrodes. (B) After 50 cycles, surface SEM images of the bare Zn electrode and the COF-h-modified Zn anode. (C) Cycling performance of the COF@Zn||MnO2 and bare Zn||MnO2 batteries at 0.5 A g-1. (A-C) Reprinted with permission from[107]. Copyright 2022, American Chemical Society; (D) Schematic diagram of the DP additive’s action mechanism on bulk electrolyte and electrode/electrolyte interfaces. (E) Cycling performance of Zn||Zn symmetric cells. (F) CE of Zn||Cu asymmetric batteries. (G) Cycling performance at 4 A g-1. (D-G) Reprinted with permission from[108]. Copyright 2025, Royal Society of Chemistry. CEI: Cathode electrolyte interphase; COF: covalent organic framework; SEI: solid electrolyte interphase; SEM: scanning electron microscopy; DP: diethyl phosphoramidate.
Constructing a 3D porous structure with high specific surface area can decrease the current density and homogenize zinc ion deposition behavior, thereby suppressing dendrite formation. Recently, Hyun et al.[109] designed and synthesized 3D-structured copper nanosheets. This material significantly enhances the wettability of zinc electrolytes, as its hydrophilic nanostructure constructs efficient transport channels that effectively promote the mass transfer process of zinc ions. Additionally, the edge surfaces of these copper nanosheets harbor uncoordinated atomic sites that exhibit exceptional zinc affinity. These zinc (002) crystal planes not only demonstrate outstanding corrosion resistance but also suppress the propagation of zinc dendrites at the root of crystal growth [Figure 9A]. The 3D Cu NS exhibits a dominant (111) crystal plane orientation. The contact angle of the copper nanosheets was significantly reduced to 10.6°, showing their pronounced hydrophilicity [Figure 9B]. Electrochemical testing revealed that the Cu NS/Zn asymmetric cell achieved a CE of 99.8% at 5 mA cm-2 and cycled stably for 1,700 cycles [Figure 9C]. When assembled into a full cell with commercial V2O5, it still cycled stability for 3,500 cycles at 5 A g-1 [Figure 9D]. Effectively resolving zinc dendrite issues while reducing zinc consumption.
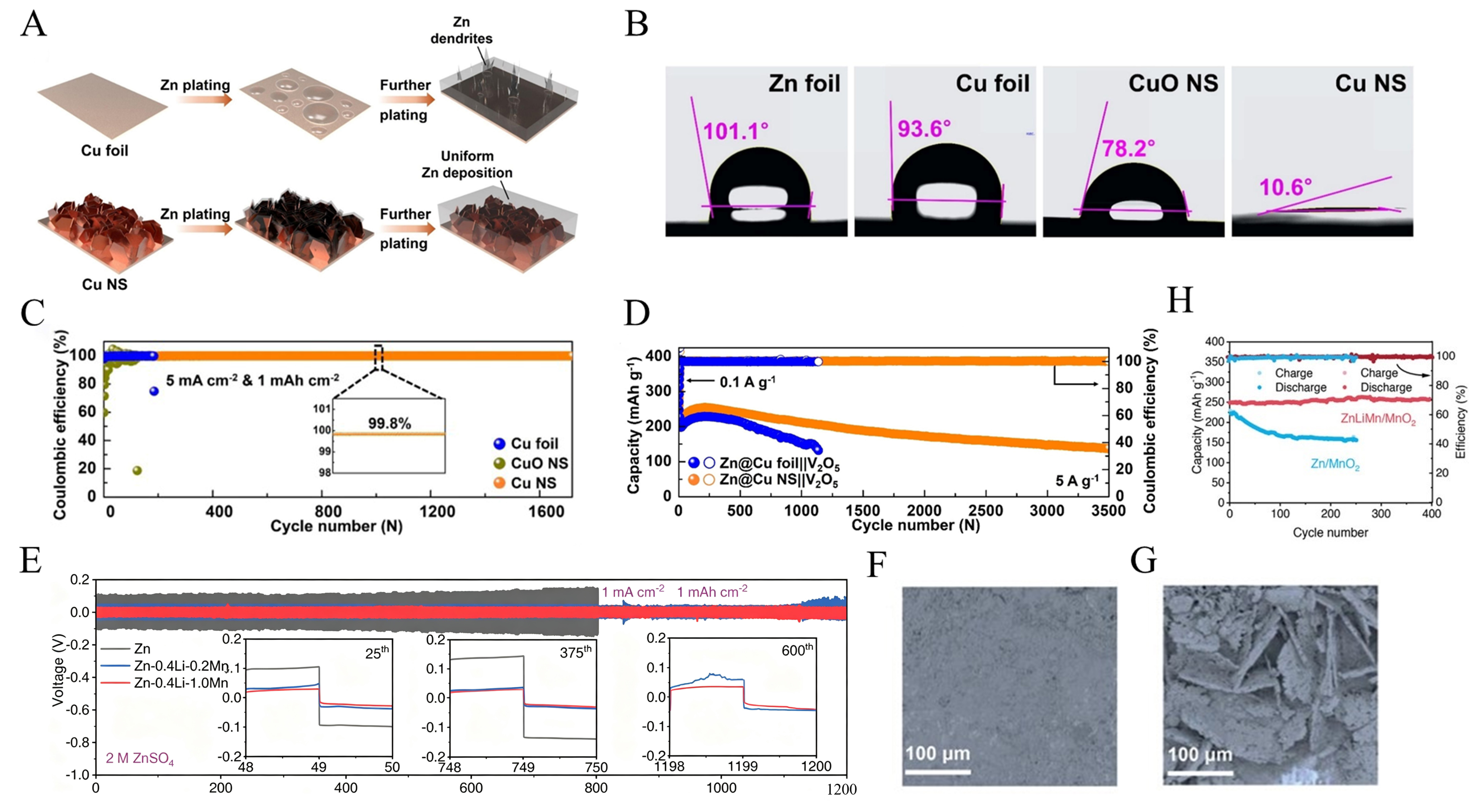
Figure 9. (A) Comparative Mechanism of Zn Deposition on Cu Foil and Cu NS. (B) Comparison of contact angles on different electrodes. (C) Electrochemical performance of asymmetric batteries based on Zn||Cu Foil, Zn||CuO NS, and Zn||Cu NS. (D) Cycling ability of Zn@Cu foil||V2O5 and Zn@Cu NS||V2O5 full cells at 5 A g-1. (A-D) Reprinted with permission from[109]. Copyright 2025, Elsevier; (E) Long-term stripping/plating cycle curves for pure Zn, Zn-0.4Li-0.2Mn, and Zn-0.4Li-1.0Mn symmetric cells. (F) and (G) are SEM images of ZnLiMn alloy and Zn after long-term stripping/galvanizing cycles, respectively. (H) Performance Comparison Between ZnLiMn Batteries and Zn/MnO2 Batteries. (E-H) Reprinted with permission from[110]. Copyright 2022, Wiley. SEM: Scanning electron microscopy.
Alloying zinc with other metals alters its crystallization behavior, thereby enhancing corrosion resistance and suppressing the HER. Zhang et al.[110] prepared a ternary Zn-Li-Mn alloy anode through minor Li, Mn, and Zn alloying. The standard electrode potentials of Li and Mn are lower than that of Zn, enabling them to preferentially react with O2 to form Li+ and Mn4+. This reduces Zn oxidation and the production of zinc-based byproducts [ZnSO4(OH)6·H2O], thereby preventing anode passivation. It maintained stable cycling performance for 1,000 h without dendrite formation after cycling [Figure 9E-G]. Zn-Li-Mn-based zinc-based batteries exhibit more stable performance, maintaining over 96% capacity retention after
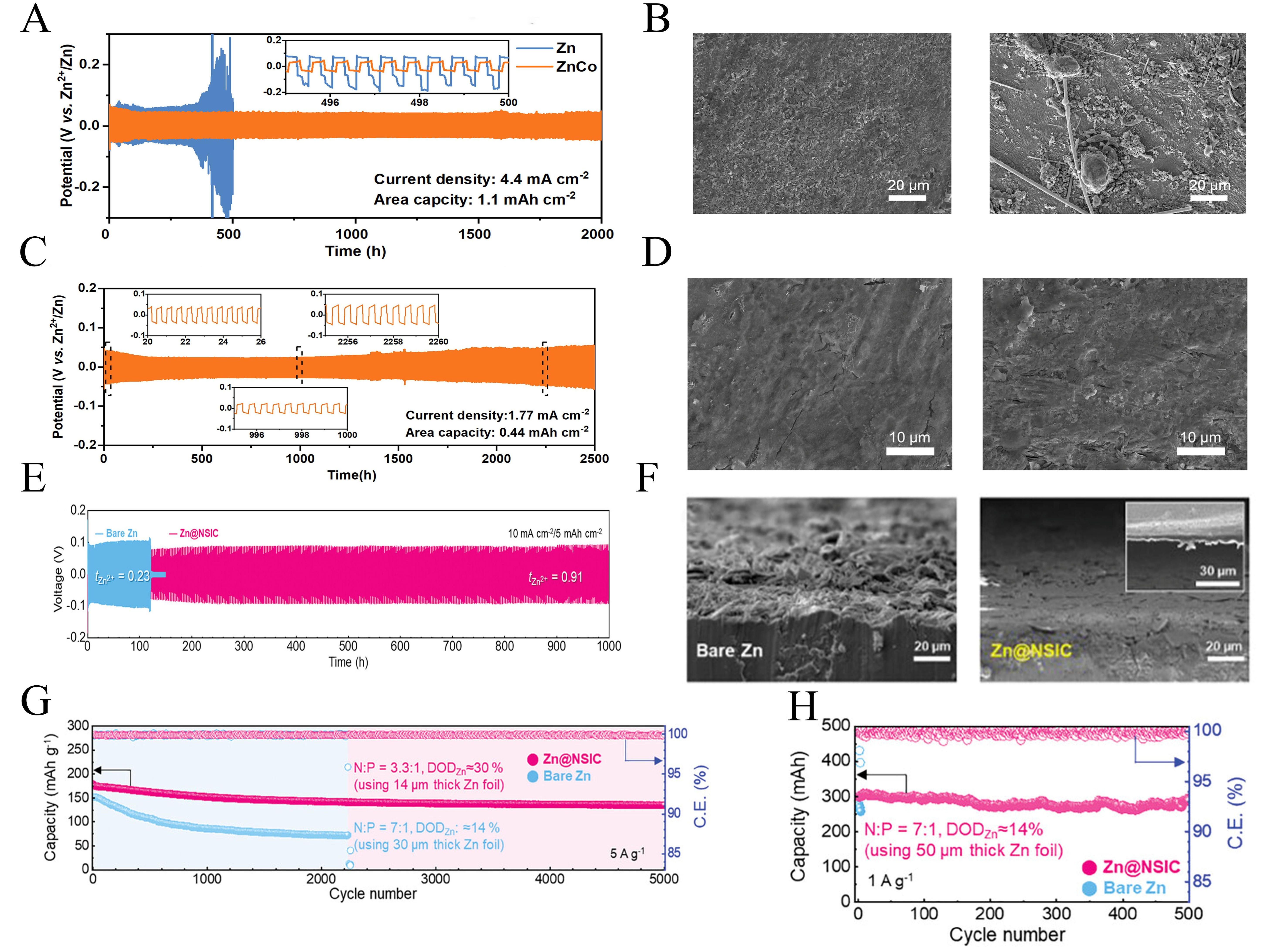
Figure 10. (A) Cycling performance of bare Zn and ZnCo symmetric cells. (B) SEM images of ZnCo and bare Zn after 50 cycles. (C) Cycling performance of ZnFe batteries. (D) SEM images of ZnFe and after 50 cycles at 1.77 mA cm-2 for 0.44 mAh cm-2. (A-D) Reprinted with permission from[111]. Copyright 2024, Elsevier; (E) Galvanostatic cycling performances of symmetrical cells with Zn@NSIC and bare Zn anodes at 10 mA cm-2. (F) Tilted SEM images of bare Zn anode and Zn@NSI Canode after cycling in symmetrical cells. (G) Cycling performances at a current density of 5 A g-1. (H) Cycling performances of large-sized full cells at 1 A g-1. (E-H) Reprinted with permission from[112]. Copyright 2025, Wiley. SEM: Scanning electron microscopy.
Forming an artificial protective layer on the surface of the zinc anode physically isolates the zinc metal from the electrolyte, suppresses side reactions, and simultaneously guides the deposition of Zn2+. Jeon et al.[112] designed a near-single-ion-conductive (NSIC) protective layer. Through the synergistic interaction of sulfonated polyether ether ketone (SPEEK) and amine-functionalized metal-organic framework (MOF) [MIL-101(Cr)-NH2]. This synergistic approach achieved a high Zn2+ migration number of 0.91 to fundamentally suppress dendrite growth. Additionally, the symmetric cell incorporating Zn@NSIC demonstrated stable performance under sustained high current density and capacity limitation for 1,000 h [Figure 10E]. Moreover, no dendrite formation was observed, the NSICl layer is coated [Figure 10F]. At
Unlike the robust and well-defined SEI in non-aqueous lithium-ion batteries, the formation of an effective SEI on zinc anodes in aqueous electrolytes remains a subject of intense debate. This limitation is primarily governed by the intrinsically narrow electrochemical stability window of water (1.23 V) and the competitive kinetics between water decomposition and the reduction of salt anions. In traditional mild aqueous electrolytes [e.g., ZnSO4, Zn(CF3SO3)2], the thermodynamic reduction potential of H2O is typically more positive than that of most salt anions. Consequently, the HER preferentially occurs at the anode surface, kinetically outcompeting the reductive decomposition of electrolyte components required for SEI formation[113,114]. Consequently, the spontaneous formation of a dense and uniform SEI film on the zinc surface is kinetically hindered. Instead, the accumulation of insulating corrosion byproducts - such as
At present, in aqueous zinc-ion batteries, the construction of a uniform and Zn2+ conducting SEI mainly relies on three types of control strategies: (1) by using “salt-in-water” type high-concentration electrolytes, solvents or functional additives to regulate the Zn2+ solvation structure, reducing the activity of free water, promoting the preferential reduction of anions such as OTf-, TFSI-, and BF4-, and forming an inorganic-organic hybrid SEI mainly composed of ZnF2, ZnS, Zn3(PO4)2 and organic polymers[115,116]; (2) a gradient zinonative artificial SEI can be in-situ constructed through electrolyte additives. The DP additive mentioned in this review can prevent the contact between the Zn negative electrode and the electrolyte while ensuring the rapid transmission of Zn2+[108]; (3) an artificial SEI protective layer can be pre-constructed on the surface of the zinc anode. Materials such as covalent organic frameworks, metal organic frameworks, and single-ion conductive polymers can be selected[117-119]. By utilizing the regular and orderly ionic channels, the interfacial side reactions can be inhibited at the physical level, while the migration behavior of zinc ions can be regulated to guide the uniform deposition of metallic zinc.
Electrolyte engineering
The electrolyte is a crucial component in batteries, serving as the primary medium that bridges the cathode and anode while facilitating ion transport. Its composition and properties directly dictate the interfacial electrochemical behavior of the zinc anode. However, ZIBs face several critical challenges. During the charge and discharge processes, the uneven deposition of Zn2+ on the anode surface induces the growth of zinc dendrites. These dendrites not only threaten battery safety by potentially penetrating the separator, but also lead to the mechanical detachment of metallic zinc. This shedding generates electrochemically inactive “dead zinc”, thereby significantly compromising the CE and cycle life of the battery[120,121]. The presence of aqueous electrolytes makes zinc anodes highly susceptible to parasitic side reactions. Specifically, the HER consumes both active materials and the electrolyte, eventually leading to severe battery swelling. Concurrently, corrosion and passivation processes generate insulating by-products that significantly increase interfacial impedance. Ultimately, these intertwined processes cause severe degradation in overall battery performance[122]. Traditional aqueous electrolytes are inherently constrained by the narrow thermodynamic decomposition voltage of water (1.23 V). This limitation restricts the selection of high-voltage cathode materials and subsequently hinders the enhancement of the battery’s overall energy density[123]. Furthermore, standard aqueous electrolytes suffer from severe performance degradation under extreme temperatures. At low temperatures, freezing readily occurs, causing a drastic reduction in ionic conductivity, whereas high temperatures exacerbate parasitic side reactions - both of which severely impede reliable battery operation[124]. Consequently, developing electrolytes capable of operating over a broad temperature range is crucial for expanding their practical applications. Kang et al.[125] designed a electrolyte by introducing the low-polarity cosolvent diethylene glycol dimethyl ether (DGM) into a zinc perchlorate [Zn(ClO4)2] aqueous solution. DGM break the hydrogen bond in H2O, regulate the freezing point of the electrolyte to -105 °C[125]. Simultaneously, it effectively suppresses dendrite growth on the zinc anode, hydrogen evolution reactions, and the formation of byproducts. In performance testing, the Zn||Zn symmetric cell demonstrated a cycle life of up to 5,200 h at 1 mA cm-2 and 1 mAh cm-2 under -20 °C conditions [Figure 11A]. Zn||polyaniline batteries achieved ultra-long cycling ability of 10,000 cycles at 1 A g-1 with no capacity decay [Figure 11B]. Dong et al.[126] constructed high-entropy electrolytes by enhancing chloride salt diversity. They utilized high mixed entropy to regulate the coordination environment, breaking the hydrogen bond of H2O and inhibiting [ZnClm]n2n-mn cluster aggregation, forming small and diverse solvated clusters. Concurrently, the excess entropy enhancement endowed the electrolyte with low-freezing properties, maintaining liquid state at
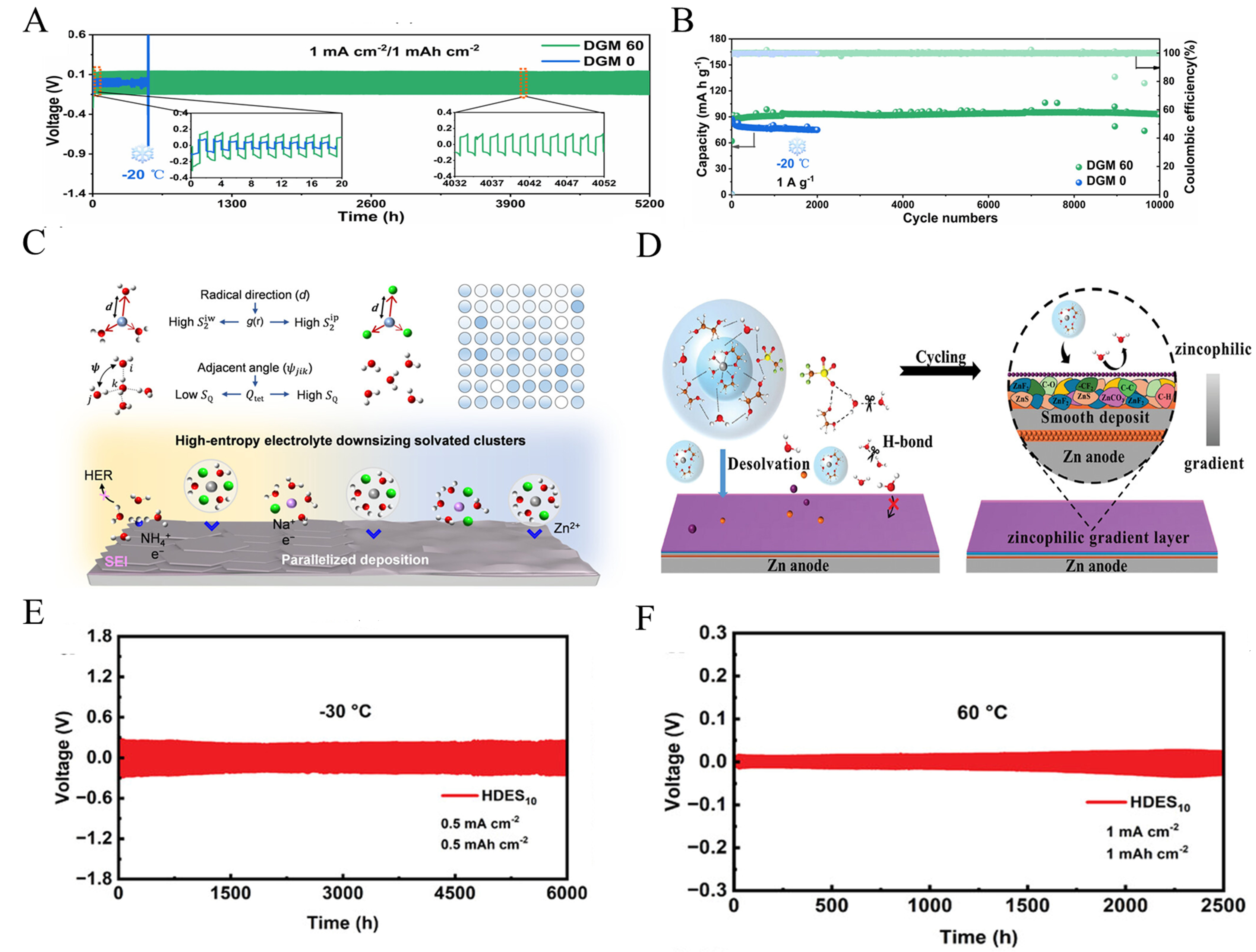
Figure 11. (A) Voltage-time curves of cells at -20 °C. (B) Cycling of Zn||PANI cells under -20 °C at 1A g-1. (A and B) Reprinted with permission from[125]. Copyright 2024, Wiley; (C) Schematic diagrams of integrating entropies in electrolytes. Reprinted with permission from[126]. Copyright 2025, American Chemical Society; (D) Schematic representation of the chemical behavior of the hydrated deep eutectic electrolyte at the bulk phase and interface. (E) Long cycle performance plots of Zn//Zn symmetric cells at -30 °C for HDES10. (F) Long cycle performance plots of Zn//Zn symmetric cells at 60 °C for HDES10. (D-F) Reprinted with permission from[127]. Copyright 2024, Wiley.
A high-quality SEI film effectively suppresses the continuous interaction between the electrode and the electrolyte, thereby preventing dendrite growth and electrode corrosion. Due to the unique nature of SEI films, the prevailing consensus is that they can only be formed in non-aqueous electrolytes. Consequently, achieving the successful formation of stable SEI films in aqueous energy storage systems still presents numerous challenges. Chen et al.[127] designed a new hydrated deep eutectic solvent (HDES)-based electrolyte. By dual confinement of free water molecules through coordination with ethylene glycol (EG) and Zn2+ and hydrogen bonding with water the activity of H2O was reduced and the electrochemical window was expanded. Simultaneously, the dissociation-reduction reaction between EG and SnI4 facilitates the formation of an organo-inorganic hybrid solid electrolyte interphase layer with a zinc-affinity gradient on the Zn surface [Figure 11D]. This electrolyte enables the zinc/zinc symmetric cell to exhibit excellent cycling performance across a wide range of temperatures [Figure 11E and F].
Introducing small amounts of functional additives into electrolytes is a cost-effective strategy for enhancing performance. Li et al.[128] proposed the use of sodium carboxymethyl cellulose (CMC) and sodium 4-aminobenzenesulfonate (SABS) as dual anionic electrolyte additives [Figure 12A]. It was firstly adsorbed onto the zinc (101) crystal plane, reconstructs the Zn2+ solvation structure, reduces water molecule coordination, and the protective layer forms on the zinc surface. In light of this, the Zn||Zn symmetric cell stably cycled for 1,800 h at 1 mA cm-2 and 1 mAh cm-2 without short-circuiting. The Zn||CMC+SABS||KMO full cell achieved a capacity retention rate of 94.5% after 1,300 cycles at 1 A g-1. Furthermore, to address potential leakage issues with liquid electrolytes and meet the demands of flexible/wearable electronics, research into quasi-solid gel electrolytes is gaining momentum. Shi et al.[129] developed a freeze-resistant hydrogel electrolyte primarily composed of zinc tetrafluoroborate [Zn(BF4)2] and polyacrylamide (PAM) [Figure 12B]. Its core mechanism leverages the high electronegativity of fluorine atoms in BF4- to form O-H…F bonds with water molecules, thereby replacing the O-H…O hydrogen bonds between H2O. This process, combined with PAM’s 3D porous network, reduces free water and disrupts ice crystal formation. The saturated Zn(BF4)2-PAM hydrogel exhibits a low glass transition temperature of -117 °C. At -70 °C, the capacity retention rate is 41%, and after 100 cycles, the capacity retention rate is 100%. Samanta et al.[130] proposed a quasi-solid ZIB system featuring a “salt-in-water” (WiS)-based molecularly crowded polymer gel electrolyte coupled with a binder-free V2O5@MnO2 cathode [Figure 12C]. The gel electrolyte (PBBZf) was prepared via thermal-initiated polymerization using Poly(ethylene glycol) methyl ether methacrylate (PEGMA) as the molecular crowding agent, BEMA as the crosslinking monomer, and benzoyl peroxide (BP) as the thermal initiator, dissolved in 3M Zn(OTf)2. This zinc-iron-nickel battery exhibits a discharge capacity of approximately 422 mAh g-1 at 0.2 A g-1. After 5,000 cycles at 10 A g-1, its capacity retention rate reaches 79.83%. It effectively suppresses zinc dendrite growth and maintains stable performance under harsh conditions.
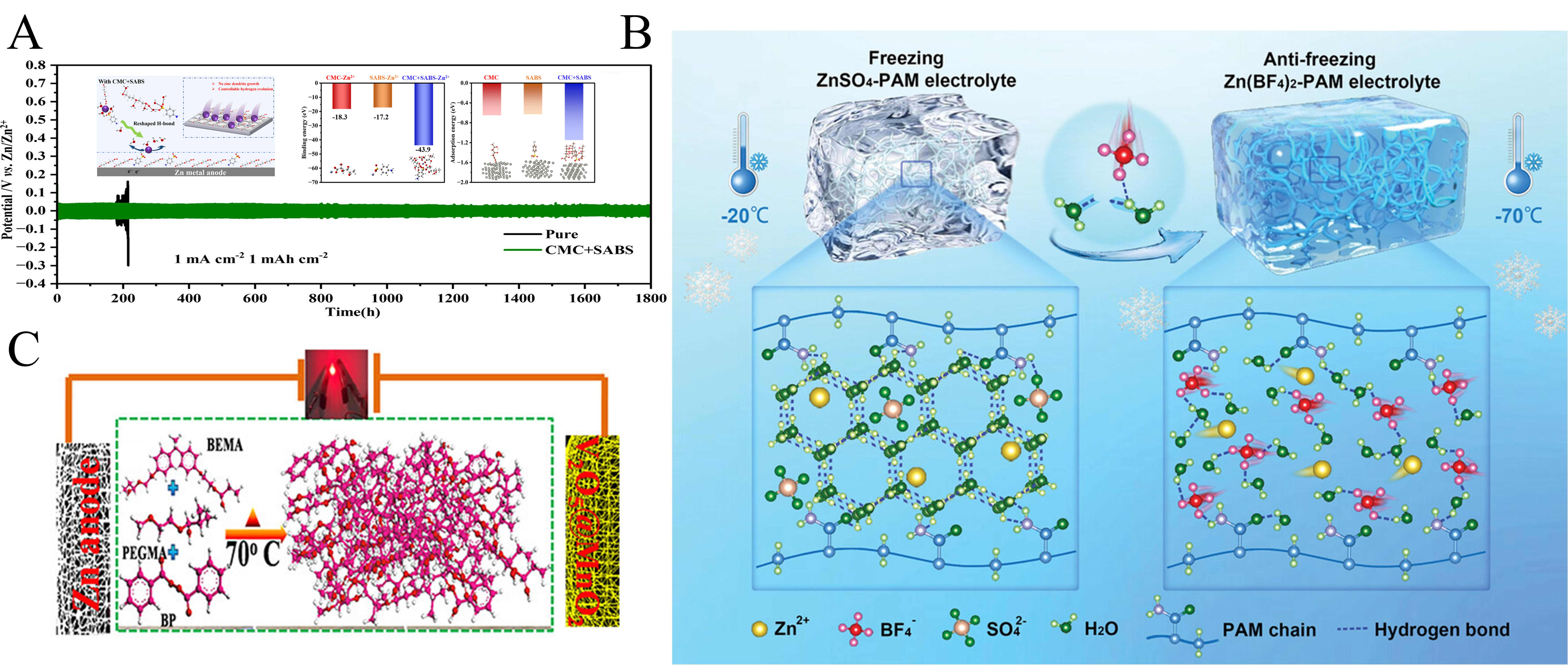
Figure 12. (A) Schematic Diagram and Performance Testing of Electrolyte Additives. Reprinted with permission from[128]. Copyright 2025, Elsevier; (B) Schematic diagram and mechanism of the hydrogel electrolytes. Reprinted with permission from[129]. Copyright 2023, Wiley; (C) Schematic diagram of a polymer gel electrolyte. Reproduced with permission: Reprinted with permission from[130]. Copyright 2022, American Chemical Society.
The decomposition of aqueous electrolytes is the core origin of interfacial side reactions, SEI/CEI structural defects and battery performance degradation, which mainly includes three pathways: (1) water splitting reaction. This is the most primary decomposition pathway of aqueous electrolytes, including the HER on the anode and the oxygen evolution reaction (OER) on the cathode. The HER will increase the local pH at the anode/electrolyte interface, inducing the formation of Zn-based hydroxide byproducts and anode passivation. The OER will accelerate the dissolution of transition metal ions in the cathode and the destruction of the CEI structure. The key to inhibiting water splitting is to reduce the activity of free water, which can be achieved by regulating the solvation structure of Zn2+ via high-concentration electrolytes, cosolvents or additives, to break the hydrogen bond network of bulk water and raise the decomposition energy barrier of water; (2) hydrolysis and decomposition of zinc salt anions. In weakly acidic aqueous electrolytes, the anions (e.g., SO42-, OTf-, TFSI-) will undergo hydrolysis reactions to varying degrees, which will change the pH of the electrolyte and aggravate the corrosion of the Zn anode[131,132]. Under high voltage, the anions will undergo oxidative decomposition on the cathode surface, and the decomposition products (e.g., sulfides, fluorides, organic carboxylates) are important components of the CEI. For example, the reductive decomposition of OTf- can form ZnF2 with high Zn2+ conductivity, which is the core inorganic component of high-quality SEI in aqueous ZIBs[133]; (3) decomposition of functional additives and cosolvents: The decomposition of organic additives/cosolvents is an important way to regulate the composition of SEI/CEI. The reductive decomposition of organic molecules on the Zn anode surface will form organic polymer components in the SEI, which can enhance the flexibility of the interfacial layer and adapt to the volume change of the electrode during cycling; while the oxidative decomposition on the cathode surface can form a compact CEI to inhibit the dissolution of active materials.
Failure mode and performance degradation analysis
ZIBs are widely regarded as highly promising candidates for large-scale energy storage due to their intrinsic safety, cost-effectiveness, and environmental benignity. However, their cycle and calendar lifespans remain constrained by complex failure mechanisms. These primarily stem from the instability of zinc anodes, the structural degradation of cathode materials, and parasitic side reactions within the electrolyte[134,135].
The failure of zinc anodes is one of the core issues leading to battery performance degradation, primarily manifested as dendrite growth, corrosion, hydrogen evolution reactions, and irreversible zinc depletion. Zinc dendrite growth is the primary failure mode causing internal short circuits in batteries, particularly pronounced under thin separator conditions[136]. Simultaneously, the zinc anode undergoes corrosion reactions in the electrolyte, which not only reduces coulombic efficiency but also continuously depletes active zinc. This ultimately leads to battery failure due to zinc depletion. Research indicates that in asymmetric batteries simulating zinc depletion conditions, zinc corrosion consumption is a more critical failure factor than dendritic short circuits[136]. Furthermore, the corrosion behavior of zinc anodes is closely linked to cathode interface reactions. For instance, the formation of pyrophosphate (ZVO) on the surface of vanadium-based cathodes disrupts the electrolyte pH, accelerating zinc anode corrosion and thereby shortening battery lifespan[137]. At extreme temperatures, the degradation process of zinc anodes is further amplified: slow zinc ion diffusion at low temperatures leads to uneven deposition, while high temperatures accelerate side reactions[138].
The structural degradation of cathode materials and the dissolution of active species constitute another significant cause of capacity decay, with the specific mechanisms varying across different material systems. For manganese-based cathodes, irreversible phase transitions are the primary driver of electrochemical failure. For example, δ-MnO2 undergoes an irreversible phase transition from a layered to a spinel structure during cycling. This process involves the rearrangement of manganese atoms, which induces a high ionic migration energy barrier, ultimately leading to deteriorated electrode kinetics and sustained capacity decay[139]. The dissolution of manganese also poses a common challenge for manganese-based cathodes, directly leading to the loss of active material. For vanadium-based cathodes, capacity decay is primarily attributed to the dissolution of vanadium species into the electrolyte and the formation of electrochemically inert by-products on the cathode surface, which impede Zn2+ insertion/extraction. In conversion-type cathodes, the shuttle effect of polysulfides, sluggish redox kinetics, and low active material utilization are the main factors limiting their cycling stability[140].
The composition and properties of the electrolyte directly influence the stability of the electrode/electrolyte interface, serving as a critical factor in mitigating failure processes. Key parameters, including pH, additives, and salt concentration, are essential for suppressing zinc dendrite formation and parasitic side reactions. For instance, even trace amounts of multifunctional additives can effectively inhibit dendrite growth and side reactions by modulating the solvation structure of zinc ions[141]. Using desolvation activation energy as a key descriptor can enable efficient screening of electrolyte additives capable of stabilizing zinc anodes[142]. Furthermore, electrolyte strategies must be tailored to accommodate extreme temperature conditions, mitigating performance degradation through optimized ion transport and interfacial reaction kinetics. Novel electrolyte systems, such as gel polymer electrolytes and Janus hydrogels, show great promise in suppressing dendrite formation and enhancing cycling stability by engineering robust interfaces and facilitating directional ion transport[143,144].
The failure of zinc-ion batteries arises from the complex interplay among the zinc anode, cathode materials, and electrolyte interfaces, with performance degradation stemming from intricate physicochemical processes. Future research must adopt a systems-level approach, employing multidimensional strategies - such as the structural design of electrode materials, electrolyte engineering, and interfacial regulation - to achieve coordinated optimization. This will enable the precise suppression of key failure pathways, thereby advancing the practical application of high-performance, long-life ZIBs.
Full battery engineering design for zinc-based batteries for practical application
Areal capacity is the key engineering parameter that distinguishes fundamental laboratory research from practical commercial devices. Commercial lithium-ion batteries for energy storage generally require a cathode areal capacity of ≥ 3 mAh cm-2. However, most reported research on ZIBs has only verified performance under ideal laboratory conditions, such as low areal capacities (< 2 mAh cm-2), ultra-thin electrodes, low active material loading, and an excess of electrolyte and zinc anode. These studies often overlook the performance degradation mechanisms and engineering optimization strategies for the full cell under practical high-areal-capacity conditions. Addressing this gap is crucial for evaluating the actual application potential of ZIBs.
Practical capacity classification and core challenges
Guided by the practical requirements of energy storage applications, we classify the areal capacity of zinc-based batteries into three tiers: (1) Laboratory-level low areal capacity (< 2 textmAh cm-2). This level is primarily utilized for fundamental cell analysis and is characterized by a low active material loading
Under practical high-capacity conditions, zinc-based all-battery systems exhibit core bottlenecks that were not evident in the laboratory half-battery tests. Under high discharge depth conditions, the failure of the zinc negative electrode becomes significantly more severe[146]. When the discharge depth exceeds 20%, the deposition of zinc ions becomes uneven, which is highly likely to trigger dendrite growth, volume expansion and corrosion problems, thereby causing internal short circuits and a sudden drop in capacity of the battery[147]. High loading levels are extremely prone to causing severe structural degradation. Thick electrodes will increase the impedance of ion transport and concentration polarization, accelerate the dissolution of Mn-based and V-based active substances, and lead to irreversible structural collapse and rapid capacity degradation. The imbalance in the matching of key parameters of the device, unreasonable N/P ratio, E/C ratio, and the design of thick electrode channels will lead to the mismatch of electrochemical kinetics between the positive and negative electrodes, accelerating the failure of the entire battery. The current distribution of large-sized battery cells is uneven. For pouch cells with an area exceeding 100 cm2, there is a tendency for local current density and electrolyte distribution to differ, which can lead to local overcharging and overdischarging, significantly reducing the battery’s cycle life and usage safety.
The full battery configuration design strategy that is adapted to and suitable for the practical capacity of the device
To address these challenges, various engineering solutions have been developed, focusing on electrode architectural design and key parameter matching. In the realm of electrode structure engineering, 3D porous current collectors (such as copper nanosheets and carbon felt) and gradient-structured thick electrodes are the prevailing strategies for mitigating the sluggish ion transport within high-loading electrodes. Shi et al.[148] developed a lightweight 3D-Cu architecture to achieve reversible zinc-metal cycling. A zinc-promoting graphene (Gr) coating was used to facilitate uniform and (002)-dominant Zn growth on the 3D-Cu surface, resulting in the formation of the 3DP-Cu@Gr structure.3DP-Cu@Gr||Zn exhibited stable performance with 700 cycles at 4 mA cm-2 and 2 mAh cm-2, with a Coulombic efficiency of > 99.6%.The zinc-loaded 3D electrode enables the symmetric battery to achieve stable cycling at 10 mA cm-2 for 300 h, providing a specific cumulative capacity of 86.7 Ah g-1. And the all-printed pantacle-shape full pouch cells (3.6 mAh) exhibit 91.4% capacity retention after 200 cycles at 1 C. In terms of the positive electrode design, constructing a vertically oriented porous structure and a gradient distribution of active substances can effectively shorten the ion diffusion distance within the thick electrode, enabling stable operation under high specific capacity conditions.
In terms of key parameter matching, the N/P ratio and the E/C ratio are the core factors influencing the total battery energy density and cycle life. An excessively high N/P ratio will reduce the energy density of the device volume, while an excessively low ratio will cause the zinc negative electrode to deplete rapidly and lead to premature failure of the battery[149]. For practical zinc-based all-battery systems, the N/P ratio needs to be adjusted to the optimal range and the E/C ratio needs to be reduced to a sufficiently low level in order to meet the core requirements of commercial applications for energy density and cost[150].
Summary and outlook
The above provides a systematic summary of ZIBs, highlighting their unique application potential in the fields of energy storage and consumer electronics. This viability stems from abundant zinc resources, low cost, high capacity, non-flammable and non-explosive aqueous electrolytes, and the use of zinc-based compounds that are low-toxicity and easily recyclable. Furthermore, this paper clearly identifies four core energy storage mechanisms: the Zn2+ (de)intercalation mechanism, the H+ and Zn2+ co-intercalation mechanism, the chemical conversion reaction mechanism, and the dissolution-deposition reaction mechanism. Detailed discussions on cathode materials, zinc anode modification, and electrolyte engineering are also provided. Collectively, these advancements provide crucial support for improving the performance and practical application of ZIBs.
Zn-based energy storage systems demonstrate considerable potential for large-scale energy storage applications, owing to their significant advantages in cost and safety. Accordingly, future research will increasingly focus on the development of high-performance zinc anodes. On the one hand, it is necessary to employ multi-scale, multi-dimensional synergistic strategies - such as combining 3D porous zincophilic frameworks with functional electrolyte additives - to thoroughly suppress dendrite growth and parasitic reactions. On the other hand, developing high-voltage, high-capacity cathode materials with structural stability is crucial for further enhancing the energy density of ZIBs, thereby broadening their application scenarios. Furthermore, advancing the large-scale production of flexible ZIBs - particularly ensuring their performance stability and cost control under extreme conditions - will be a crucial step in transitioning them from the laboratory to the marketplace. By overcoming key material and technological bottlenecks, ZIBs are poised for large-scale commercialization in applications such as stationary energy storage, consumer electronics, and wearable devices.
MAGNESIUM-BASED ENERGY STORAGE SYSTEM
Introduction to Mg-ion batteries
As a core component of advanced energy storage systems, magnesium-based materials boast three primary advantages. First, the theoretical volumetric capacity of Mg metal reaches 3,833 mAh cm-3, substantially exceeding that of Li metal (2,046 mAh cm-3). Furthermore, its standard reduction potential is -2.37 V (vs. SHE), laying a solid foundation for high energy density[151-155]. Second, the natural abundance of magnesium is approximately 104 times that of Li. This ensures low raw material acquisition costs and widespread distribution, avoiding the supply chain risks caused by the scarcity of lithium resources[156]. Third, magnesium metal typically does not form dendrites during the plating/stripping process. Compared to lithium metal anodes, which are prone to forming dendrites, this characteristic significantly reduces the risk of short circuits and enhances overall battery stability[151-155,157-159].
The core technical advantages of magnesium-based batteries are more prominent compared with similar technologies such as LIBs and Sodium-ion Batteries (SIBs). First, based on the Mg2+ double-electron transfer mechanism, the theoretical energy density is higher[160]. Second, electrolyte compatibility has been gradually improved. By designing non-nucleophilic electrolytes [Figure 13] and solid electrolytes, the corrosion problem between traditional electrolytes and magnesium anodes has been solved, while achieving matching with high-capacity cathodes[161-167]. Third, the cycle stability has been significantly enhanced, and Chevrel phase cathodes retain over 85% of their capacity after 2,000 cycles [Figure 14][37,168-170], paving the way for large-scale energy storage applications.
Figure 13. (A) Oak Ridge thermal ellipsoid plot of the molecular structure of [Mg4Cl6(DME)6][B(HFP)4]2; (B) Cycling behavior of Mg-Mo6S8 batteries with 0.5 M OMBB as electrolyte. (A and B) Reprinted with permission from[167]. Copyright 2017, Royal Society of Chemistry.
Figure 14. (A) A crystal model of Chevrel phase (CP) Mo6S8 viewed along the [211] direction, featuring three types of highly symmetric sites: 3a, 3b, and 9d. (B) Sublattice corresponding to sites 3a and 3b. (C) The inner and outer rings can hop between partially occupied external and internal sites. (A-C) Reprinted with permission from[170]. Copyright 2017, American Chemical Society; (D) Schematic Diagram of Solid-State Diffusion of Mg2+ in CP. Reprinted with permission from[37]. Copyright 2013, Royal Society of Chemistry.
Energy storage mechanism
The core working mechanism of MIBs relies on the reversible shuttling of Mg2+ ions and the associated redox reactions of the cathode host materials. During operation, this process can be summarized as the coupled interplay of ion migration, electron transfer, and host structure redox.
During discharge, metallic magnesium at the anode undergoes oxidation (Mg → Mg2+ + 2e-), releasing Mg2+ ions into the electrolyte and electrons into the external circuit. These Mg2+ ions migrate to the cathode, where they insert into the host lattice while the incoming electrons reduce the host material to maintain charge balance - this cathode process is not the reduction of Mg2+ to metallic Mg, but rather the reduction of the host material coupled with Mg2+ intercalation. The overall cathode reaction depends on the host material: for insertion-type cathodes (e.g., Chevrel phase Mo6S8) the reaction is Mo6S8 + xMg2+ + 2xe- ⇌ MgₓMo6S8[171,172]. For conversion-type cathodes (e.g., metal sulfide MS) it is MS + Mg2+ + 2e- ⇌ M + MgS[173]. And for organic cathodes with carbonyl groups, it is C=O + Mg2+ + 2e- ⇌ C-O-Mg. During charge, the process is fully reversed: Mg2+ ions de-intercalate from the cathode, migrate back, and are reduced and deposited as metallic Mg at the anode. This “rocking-chair” ion shuttling mechanism, combined with the two-electron transfer per Mg2+ ion, provides the foundation for high-capacity energy storage in MIBs.
The core feature of this mechanism is the reversible (de)intercalation or conversion of Mg2+ within the cathode host structure, accompanied by the redox reaction of the host material. This “rocking-chair” ion shuttling, along with the divalent nature of Mg2+ which enables a two-electron transfer per ion, provides the fundamental basis for the high capacity in MIBs[174,175].
Key materials for Mg-ion batteries
Cathode materials Mg-ion batteries
Cathode materials dictate the energy density of MIBs. Their development has primarily focused on lowering Mg2+ diffusion barriers, enhancing redox activity, and improving cycling stability. Currently, these cathode materials can be broadly categorized into three types: intercalation-type, conversion-type, and organic cathodes[176].
The Chevrel phase was the first cathode family to achieve reversible Mg2+ insertion, laying the foundation of magnesium-ion battery research. Yoo et al. reported layered TiS2, which was initially pillared by PY14+ to achieve an interlayer spacing of 1.1 nm (10.9 Å); this spacing was further expanded to 18.6 Å after subsequent MgCl+ insertion. This structure enables rapid diffusion of MgCl+ (rather than bare Mg2+), providing a reversible capacity of 400 mAh g-1 at 60 °C[177]. Liang et al. designed graphene-based MoS2 (G-MoS2) composites with a monolayer architecture to shorten Mg2+ diffusion pathways, achieving a reversible capacity of 170 mAh g-1 at 1.8 V[178]. The performance of V2O5-based materials has been further improved through composite engineering. For instance, in the RFC/V2O5 composite cathode reported by Cheng et al., resorcinol and formaldehyde as precursors (referred as RFC) featuring a high specific surface area of
Conversion-type cathodes achieve high capacities via redox reactions between metal cations and Mg2+. Zhang et al. first confirmed the conversion reaction mechanism of α-MnO2, the MnO6 octahedra decompose into an amorphous shell containing Mn2+ during magnesiation, although structural collapse compromises cycling stability[183]. Nam et al. exploited crystal water in birnessite-type MnO2 to enhance ion diffusion; in aqueous electrolytes the material retained 62.5% of its capacity after 10,000 cycles[184]. Sulfide conversion cathodes show even better performance: Shen et al. prepared hollow CuS nanocubes undergoing reversible Cu2S/Cu0 redox, delivering 200 mAh g-1 at 100 mA g-1 with no decay over 50 cycles[185]. Xiong et al. achieved
Organic cathodes offer abundant resources and structural tunability. Sano et al. showed that 2,5-dimethoxy-1,4-benzoquinone (DMBQ) coordinates Mg2+ at carbonyl sites, delivering 260 mAh g-1 at
In non-aqueous magnesium-ion batteries, the formation of the CEI and the surface reconstruction of the cathode materials are the key factors limiting cycle stability and rate performance. The primary driving forces for these phenomena are the strong Coulombic interactions between the high-charge-density Mg2+ ions and the host lattice, coupled with the oxidative decomposition of the electrolyte at high voltages.
For intercalation-type cathodes, the repeated insertion and extraction of high-charge-density Mg2+ ions will induce significant lattice stress and volume changes, leading to the irreversible amorphization of the material surface and structural reconstruction[191]. For instance, during the magnesium insertion process, layered MnO2 undergoes irreversible phase transformation and structural collapse, accompanied by the dissolution of Mn ions. This is the core reason for the rapid capacity degradation of manganese-based oxide cathodes in non-aqueous magnesium-ion batteries. For conversion-type cathodes (such as CuS, NiS2), the conversion reaction leads to the pulverization of the active material, forming non-uniform CEI on the surface, ultimately causing the shuttle effect of polysulfides and the loss of active materials[186,192-194].
In aqueous systems, the oxidative decomposition of the electrolyte on the positive electrode surface constitutes the primary source of CEI components. For high-voltage oxide cathodes, nucleophilic groups in the electrolyte readily react with the electrophilic high-valent transition metal ions on the cathode surface, leading to continuous electrolyte decomposition. This results in the formation of a thick, ionically insulating CEI layer on the cathode surface, which significantly increases the interfacial impedance and hinders Mg2+ diffusion. The design of non-precious-metal-based electrolytes incorporating high-voltage-stable magnesium salts represents the core solution to this challenge. A boron-centered magnesium salt electrolyte with an anodic stability of up to 4.3 V can form a uniform and ultra-thin CEI on the positive electrode surface, which not only suppresses the continuous decomposition of the electrolyte but also facilitates rapid Mg2+ transport. In addition, surface coating of the positive electrode material can prevent direct contact between the active material and the electrolyte, inhibit detrimental surface reconstruction, and regulate the formation of a high-quality CEI.
Anode materials Mg-ion batteries
The early stage of pure-magnesium anodes was characterized by the coexistence of advantages and limitations. As early as 2000, pure magnesium metal became the preferred anode material for MIBs at that time, due to its elevated volumetric capacity and dendrite-free characteristic[195]. Aurbach et al. first paired a pure magnesium foil anode with a Chevrel-phase Mo3S4 cathode to construct the first complete and practical rechargeable magnesium battery prototype. At 100% depth of discharge, the prototype achieved over 2,000 cycles with less than 15% capacity decay[151]. However, pure-magnesium anodes have two key issues: (i) they react with most electrolytes to form a passivation layer, which hinders Mg2+ transport and results in a CE below 80%[196,197]; (ii) the chemical diffusion coefficient of Mg2+ is low in many cathode lattices, leading to pronounced polarization at high current rates[198].
In order to surmount the inherent limitations of pure-magnesium anodes in passivation and volumetric stability, alloying emerged as a core strategy to break performance bottlenecks. Wu et al. reported spinel Li4Ti5O12 (LTO) as a Mg2+ insertion anode; its low-strain feature (volume change < 1%) enabled a reversible capacity of 175 mAh g-1 with no obvious decay over 500 cycles[199]. Zeng et al. prepared mesoporous Li3VO4/C (LVO/C) hollow spheres via spray-drying. The carbon coating effectively enhances electronic conductivity, delivering an initial discharge capacity of 318 mAh g-1 and avoiding the passivation issue associated with pure magnesium anodes[200]. Alloying anodes further evolved to intermetallic compounds[201]. Niu et al. developed nanoporous Bi6Sn4 alloys, where a biphase synergistic effect mitigates volume expansion; the material delivered 434 mAh g-1 at 50 mA g-1 and retained 280 mAh g-1 after 200 cycles at 200 mA g-1[175]. Such materials replace simple deposition of pure magnesium with alloying reactions, effectively suppressing formation of passivation layer and improving diffusion kinetics.
Subsequently, emerging composite anodes brought two-dimensional materials and artificial interfacial modification to the forefront. Majid et al. showed that a bilayer Al2CO structure used as an anode could deliver a theoretical capacity of 1,634.73 mAh g-1[202]. Tasawar et al. reported layered InOCl anodes with a large theoretical capacity of 4,700 mAh g-1 and a magnesiation-induced volume expansion of only 3.6%, indicating promising structural stability[203]. Artificial interfacial engineering further alleviates interfacial kinetic bottlenecks. Zhang et al. constructed a Zn/ZnCl2/MgZn2/MgCl2 artificial interface via a ZnCl2-Mg metal displacement reaction, which effectively resolved the compatibility issues between Mg(TFSI)2-based electrolytes and magnesium anodes, reducing the Mg plating or stripping overpotential to ~0.2 V and accelerating Mg2+ transport[204]. Wei et al. coated the Mg anode with liquid gallium to form a Ga5Mg2 alloy interfacial layer; this layer contains active Ga sites that lower the nucleation energy barrier and enable more stable Mg deposition and cycling[205].
The formation of SEI on the Mg anode in non-aqueous electrolytes and the accompanying passivation issues are the most critical interface challenges in magnesium-ion batteries, which are fundamentally different from aqueous zinc-ion batteries. While metallic Li can form a Li+-conductive SEI in most organic electrolytes, metallic Mg reacts with traditional carbonate- or ether-based solvents, as well as conventional magnesium salts such as Mg(TFSI)2 and Mg(ClO4)2, to form a dense and ion-insulating passivation layer on its surface. This passivation layer is mainly composed of MgO, Mg(OH)2, MgCO3, and magnesium alkoxides. Its diffusion coefficient for Mg2+ is extremely low, which completely hinders the reversible plating and stripping of Mg, resulting in a sharp increase in interfacial impedance and even the complete loss of electrochemical activity at the Mg anode.
To construct a high-quality SEI with satisfactory Mg2+ conductivity in non-aqueous electrolytes, the key lies in regulating the reduction and decomposition pathways of the electrolyte to prevent the formation of ion-insulating magnesium-based compounds. The development of noble-metal-free electrolytes represents a milestone in solving the problem of Mg anode passivation. The APC electrolyte and magnesium borate-based electrolyte, whose reduction decomposition products are Mg-Cl complexes with high Mg2+ electrical conductivity, can form stable and ion-permeable SEI, and will not form a passivation layer on the Mg surface[206]. In recent years, significant progress has been made in regulating the SEI through the use of electrolyte additives. Additives such as Mg(BH4)2 and GeCl4 can undergo preferential reduction on the Mg surface, forming an Mg-B- or Ge-based SEI. This process effectively removes the native passivation layer and stabilizes the Mg plating and stripping process[207,208].
Furthermore, artificial interface engineering is an effective strategy for addressing the passivation issue of the Mg anode. Specifically, the Zn/ZnCl2/MgZn2/MgCl2 artificial interface and the Ga5Mg2 alloy interphase layer can prevent direct contact between the Mg anode and the electrolyte. This avoids the formation of an ion-insulating layer, while simultaneously reducing the nucleation energy barrier for Mg plating and ensuring the rapid transport of Mg2+ ions[204]. Unlike the core goal of SEI regulation in aqueous zinc-ion batteries (suppressing dendrite growth and HER), the core of SEI regulation in non-aqueous magnesium-ion batteries is to solve the Mg electrode passivation problem and achieve reversible deposition/solubilization of Mg.
Electrolyte engineering
Serving as the transport medium for Mg2+, electrolytes must simultaneously deliver high ionic conductivity, a broad electrochemical stability window, and excellent interfacial compatibility with electrodes. Broadly speaking, these electrolytes can be classified into liquid and solid-state systems.
Liquid electrolytes have evolved from nucleophilic to non-nucleophilic systems. In 1990, Gregory et al. first reported Grignard-reagent-based electrolytes that enabled reversible Mg plating/stripping, albeit with a narrow electrochemical stability window[209]. In 2008, Mizrahi et al. developed the APC electrolyte [(PhMgCl)2-AlCl3/THF], which enabled highly reversible Mg plating and stripping. Featuring a stable electrochemical window and superior ionic conductivity, it emerged as an early benchmark electrolyte for magnesium batteries[206]. Because APC contains nucleophilic species (e.g., PhMgCl) and is corrosive to non-noble metals, it corrodes current collectors and reacts with electrophilic cathodes, severely limiting its practical use in full cells[210]. Ionic liquids and mixed-solvent electrolytes were subsequently explored to address corrosivity. Zhao-Karger et al. formulated a (HMDS)2Mg-AlCl3/G4 [hexamethyldisilazide (HMDS)] electrolyte; by tuning the solvation structure of Mg2+ with an ionic liquid, the electrolyte showed improved compatibility with sulfur cathodes and delivered 260 mAh g-1 over 20 cycles[164]. Since 2020, non-nucleophilic electrolytes have become key matches for high-voltage cathodes. Zhang et al.[211] designed a boron-centered anion-based magnesium electrolyte (BCM electrolyte) prepared in 1,2 dimethoxyethane (DME) via a one-pot formulation of tris(2H-hexafluoroisopropyl) borate (THFPB) and MgF2. After composition optimization and electrochemical conditioning, it exhibited an ionic conductivity of 1.1 mS cm-1 and a Coulombic efficiency of 99.8% at the 50th cycle. Furthermore, based on the 5.3 V anodic stability measured vs. Li+/Li in the Li system and an empirical Li-Mg conversion, the upper limit of anodic stability for Mg2+/Mg was estimated to be approximately 4.3 V.
Solid-state electrolytes (SSEs) have garnered significant attention since 2018 due to their intrinsic safety and leak-free characteristics. Notably, Hassan et al. developed MOF-based solid electrolytes (MOF-SEs). The structural disorder inherent in amorphous Mg-bp3dc provides additional diffusion pathways; consequently, at 30 °C, it achieved an ionic conductivity of 3.8 × 10-5 mS cm-1 and a Mg2+ transference number of 0.335[212].
Electrolyte additives serve as a vital auxiliary strategy to optimize the electrode-electrolyte interface (EEI), thereby enhancing overall electrochemical performance. Li et al. added Mg(BH4)2 to Mg[B(hfip)4]2/DME, removing the passivation layer on the Mg anode and stabilizing the plating/stripping overpotential after cycling, which was reduced by 50 mV compared with the blank electrolyte-significantly improving the kinetics of Mg deposition/dissolution[208]. Zhang et al. introduced GeCl4 to Mg(TFSI)2/DME to form an in-situ Ge-based protection layer; symmetric cells cycled for 1000 hour without obvious decay[207]. Wang et al. used a Pyr14+ additive to electrostatically guide lateral Mg2+ deposition, suppress electrolyte decomposition and interfacial side reactions, and achieve a capacity of 50 mAh g-1 over 40 cycles with V2O5 cathodes[213].
In stark contrast to aqueous systems, the decomposition of non-aqueous electrolytes is the fundamental cause of Mg anode passivation, structural defects in the CEI, and interfacial side reactions in MIBs. It mainly involves three major pathways: (1) Reduction decomposition of organic solvents occurs on the surface of the Mg negative electrode. Traditional carbonate-based solvents [such as ethylene carbonate (EC), dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC)] undergo reductive decomposition on the highly reducing Mg surface, generating MgCO3, Mg(OH)2, and magnesium alkoxides, which are the main components of the ionically non-conductive passivation layer. The reductive stability of ether-based solvents (such as THF and DME) is higher than that of carbonate-based solvents[214,215]. Therefore, ether-based solvents are currently the mainstream choice for magnesium-ion battery electrolytes. However, ether-based solvents can still undergo ring-opening decomposition catalyzed by trace amounts of water and impurities, eventually leading to the formation of a passivation layer on the Mg surface; (2) The decomposition of magnesium salt anions is a key factor determining the composition of SEI/CEI. For traditional magnesium salts such as Mg(TFSI)2 and Mg(ClO4)2, the TFSI- and ClO4- anions will be reduced and decomposed on the Mg surface, generating insulating compounds such as MgF2 and MgO, which leads to negative electrode passivation[216-218]. Conversely, the anions such as [B (hfip)4]- and AlCl4- in non-protic electrolytes do not form insulating magnesium-based compounds after decomposition, but instead form Mg-Cl and Mg-B complexes with high Mg2+ conductivity. This is the core reason why these electrolytes can achieve reversible deposition/solubilization of Mg; (3) Nucleophilic-electrophilic side reactions occur between electrolyte components and electrodes. In traditional APC electrolytes, the strong nucleophilicity of Grignard reagents induces reactions with electrophilic high-voltage oxide cathodes and current collectors, leading to electrolyte decomposition and cathode corrosion. Consequently, non-nucleophilic electrolytes have emerged as the predominant choice in MIBs research. Furthermore, trace amounts of water and oxygen within the electrolyte react with the highly active Mg metal to form passivation products such as MgO and Mg(OH)2, which significantly accelerate anode passivation. Therefore, rigorous control of moisture and oxygen levels is a fundamental prerequisite for the stable operation of non-aqueous magnesium-ion batteries.
Failure mode and performance degradation analysis
MIBs are considered promising candidates for next-generation energy storage technologies due to their high theoretical energy density, cost-effectiveness, and inherent safety. However, their practical application remains impeded by rapid performance degradation caused by multiple failure mechanisms. These failures primarily stem from the high charge density of Mg2+, which induces sluggish diffusion kinetics within the host electrode materials and triggers complex interfacial side reactions[219,220]. Ultimately, this performance degradation is the result of the complex interplay among electrode materials, electrolytes, and their interfaces, which persists throughout the entire electrochemical cycling process.
The degradation of cathode materials represents one of the primary bottlenecks in the performance decline of MIBs. For traditional intercalation-type cathode materials, such as spinel manganese oxides, the strong Coulombic interactions between Mg2+ and the host lattice induce sluggish solid-state diffusion kinetics and severe structural strain. Consequently, this leads to pronounced voltage hysteresis and rapid capacity fading. For example, spinel manganese oxide cycled in conventional electrolytes exhibits significant polarization and rapid capacity decay[221]. In contrast, conversion-type systems (such as those in magnesium-sulfur batteries), while thermodynamically more favorable, face challenges including the dissolution and shuttle effect of magnesium polysulfides, passivation of the sulfur cathode, and sluggish conversion kinetics. These factors collectively lead to irreversible active material loss and low Coulombic efficiency[222-224]. Nano-structured designs, such as the use of nanoscale ζ-V2O5, have been demonstrated to enhance the reversible storage capacity of Mg2+ ions and alleviate kinetic limitations[225].
The degradation of the magnesium anode is primarily attributed to the non-uniform deposition/dissolution of metallic magnesium and the formation of an unstable electrode/electrolyte interface (EEI). In liquid electrolytes, a passivation layer readily develops on the magnesium surface, which impedes the reversible plating/stripping of Mg2+ ions. Consequently, this phenomenon leads to elevated overpotentials and a significantly compromised cycle life[226]. Alloy anodes (such as InSb and MgBi) can mitigate passivation to some extent, but their interface reactions with electrolytes are complex. Research indicates that different electrolyte systems form interface layers with vastly different compositions and properties on the alloy surface, directly affecting magnesium ion transport kinetics and cycling stability. In solid electrolytes, the MgBi alloy surface forms a bubbly, unstable SEI, leading to uneven magnesium deposition. Adding electrolyte additives can suppress this SEI formation, thereby significantly improving cycle performance.
At the full battery system level, performance degradation results from the coupling of multiple factors. First, electrolyte compatibility is a critical challenge. Electrolytes that are compatible with magnesium metal anodes are often incompatible with high-voltage oxide cathodes, and vice versa. This incompatibility severely restricts material selection and may induce undesirable side reactions[219]. Second, irreversible structural evolution across multiple scales, encompassing cathode material degradation, dendrite growth on anodes, and continuous interfacial phase evolution, collectively leads to increased internal resistance and irreversible capacity loss. Advanced in situ characterization techniques provide powerful tools for the real-time observation of these degradation processes[227-229]. Finally, in systems such as magnesium-sulfur batteries, the shuttle effect of magnesium polysulfides not only leads to active material loss but may also corrode the anode, accelerating overall failure[230]. The failure and performance degradation of magnesium-ion batteries represent an intricate, multiscale challenge spanning bulk electrode structures, interfacial electrochemistry, and system-level compatibility. This primarily originates from the pronounced polarization of Mg2+, which engenders sluggish solid-state diffusion within cathode materials and induces interfacial instability at the anode. Future research should synergistically integrate nanoscale material engineering, precise interfacial control, electrolyte innovation, and multiscale in situ characterization to thoroughly elucidate and effectively suppress key degradation mechanisms, thereby accelerating the development of high-performance MIBs.
Engineering design for practical application of Mg-based batteries full battery engineering
Unlike zinc-based batteries, magnesium-based full-cell systems face more severe engineering challenges in achieving high practical specific capacity. This is primarily due to the slow diffusion kinetics of Mg2+ within thick, high-loading cathodes, which leads to pronounced voltage hysteresis and low utilization of active materials. Furthermore, at high depths of discharge, significant passivation of the magnesium anode occurs, and the deposition/stripping overpotential continuously increases, resulting in rapid capacity degradation. Moreover, the non-nucleophilic electrolytes typically used in these systems exhibit poor compatibility with high-loading oxide cathodes, triggering continuous side reactions and electrolyte consumption. The high cost of these electrolytes, combined with the stringent requirement to strictly control the E/C ratio, further exacerbates the interface stability issues of the system.
Currently, research on magnesium-based full-cell systems aiming for practical capacities predominantly focuses on two categories of cathodes: intercalation-type and conversion-type. Among intercalation-type materials, the Chevrel phase Mo6S8 is the most technologically mature cathode. When assembled with a magnesium foil anode and an APC electrolyte, the resulting full cell delivers a stable areal capacity of
Regarding innovations in full-cell architecture, the anode-free magnesium full cell has emerged as a prominent research hotspot in recent years due to its potential to significantly enhance the volumetric energy density of the device[231]. By pre-depositing metallic magnesium onto a copper current collector and pairing it with a high-capacity Mo6S8 cathode, the anode-free full cell achieved an areal capacity of 2.8 mAh cm-2, delivering a 42% increase in volumetric energy density compared to conventional full cells utilizing a magnesium foil anode. Furthermore, the development of bipolar architectures and solid-state full cells provides novel strategies for enhancing the energy density and safety of MIBs.
However, it should be noted that currently reported magnesium-based full cells still generally rely on a large excess of magnesium at the anode (N/P ratio > 5) and high electrolyte volumes (E/C ratio > 50 μL mAh-1). Research on full cells operating under lean-electrolyte and low N/P ratio conditions remains exceedingly scarce. Furthermore, achieving long-term cycling stability in large-format magnesium pouch cells at practical capacities remains a critical challenge that has yet to be overcome.
Summary and outlook
With its three core advantages, MIBs has become a highly promising energy storage technology. First, the theoretical volumetric capacity of Mg is higher than of Li, and its standard reduction potential is as low as -2.37 V (vs. SHE), laying a solid foundation for high energy density. Second, the abundance of Mg is 2.9%, approximately 104 times more abundant than Li, which not only reduces raw material acquisition costs but also avoids supply chain risks caused by the scarcity of lithium resources. Third, magnesium metal does not form dendrites during the deposition/dissolution process, greatly enhancing battery safety. The core working mechanism of MIBs is the reversible shuttling and redox reactions of Mg2+ between the cathode and anode, realizing energy conversion through intercalation/deintercalation or alloying/dealloying processes. Furthermore, the two-electron transfer characteristic further supports high-capacity output. In the field of materials, anodes have evolved from pure magnesium to alloy systems and two-dimensional composite systems, effectively addressing issues of passivation and ion diffusion. Cathodes cover insertion-type, conversion-type, and organic materials, achieving continuous breakthroughs in capacity and stability. Electrolytes have advanced from nucleophilic systems to non-nucleophilic systems and solid-state electrolytes, with the EEI optimized through additives. Compared with LIBs and SIBs, the comprehensive advantages of MIBs in energy density, cost, and safety, along with breakthroughs in existing material and mechanism research, have laid a crucial foundation for their large-scale application.
Despite their promise, the challenges currently faced by MIBs require urgent attention. First, the chemical diffusion coefficient of Mg2+ in many cathode materials is relatively low, leading to pronounced electrochemical polarization at high current rates. Second, the compatibility between high-voltage cathodes and electrolytes still needs significant optimization. Finally, the long-term cycling stability of full cells has not yet met practical application requirements. Future research should focus on three primary directions. First, the development of new functional materials - such as composite electrodes with high ionic conductivity and non-corrosive solid-state electrolytes with a wide electrochemical window - is essential to enhance ion transport efficiency and interface stability. Second, interface regulation technologies must be deepened; through the precise design of artificial interphases or multifunctional additives, side reactions can be suppressed and the overpotential of Mg plating/stripping reduced. Third, large-scale manufacturing processes need to be advanced to bridge the gap between laboratory-scale synthesis and industrial production. With the resolution of these bottlenecks, MIBs are expected to achieve widespread industrial application in the energy storage sector. The interdisciplinary collaboration among materials chemistry, electrochemistry, and engineering will be the key driving force in transitioning MIBs from theoretical research to practical deployment.
ENGINEERING OF ZINC/MAGNESIUM ION BATTERY ELECTROLYTES AND COMPARATIVE ANALYSIS OF INTERFACE STABILITY
To break through the simplistic parallel comparison approach, this paper has developed a critical analysis framework to conduct a comparative analysis of the electrolyte and interface design concepts of ZIBs and MIBs [Table 2]. In terms of common technology strategies, both systems achieve interface stability optimization through three core paths, and the technical experience has extremely strong transferability across systems: (1) The regulation of the solvation structure of divalent cations reduces the de-solvation energy barrier at the electrode/electrolyte interface, and simultaneously inhibits the side reactions caused by solvent decomposition. For instance, the co-solvent strategy widely used in zinc-ion batteries can break the hydrogen bond network of water molecules and reduce the activity of free water. This idea has also been applied in MIBs to optimize the Mg2+ solvation shell and reduce the passivation tendency of the magnesium anode; (2) Functional additive engineering involves using additives to uniformly distribute ion fluxes on the electrode surface, or to induce the formation of stable ion-conductive interface membranes. The mechanism of regulating cation deposition behavior in zinc-ion batteries through anion additives has been demonstrated to be effective in preventing uneven magnesium deposition in magnesium-ion batteries; (3) Construction of the artificial interface layer: By physically isolating the electrodes from the electrolyte, it prevents the side reactions caused by direct contact between the two, and at the same time guides the uniform deposition of ions.
Comparative analysis of electrolyte compatibility and interfacial stability in ZIBs and MIBs
| Comparison dimension | ZIBs | MIBs |
| Mainstream electrolyte system | Aquatic electrolyte, WISEs, aqueous/non-aqueous liquid electrolyte, gel electrolyte, quasi-solid/fully-solid electrolyte | Non-aqueous liquid electrolyte, gel/semisolid electrolyte, solid electrolyte, aquatic electrolyte |
| Core design principles of the electrolyte | Prioritize the suppression of water-induced side reactions (HER, corrosion), regulate the solvation structure of Zn2+, and expand the electrochemical window; also take into account the ionic conductivity, safety and cost[232,233] | Prioritize the compatibility with the metal magnesium anode (to avoid passivation), achieve reversible deposition/solubilization of magnesium, and simultaneously possess high anode stability to match the high-voltage positive electrode; balance the kinetics of Mg2+ transport and oxidation stability[234,235] |
| Interface chemical complexity | Regulate the activity of water molecules, inhibit HER, OER and corrosion by-products | Eliminate the nucleophilic corrosiveness of traditional electrolytes and break down the ionic insulating passivation layer on the magnesium negative electrode surface |
| Comparison of SEI Membrane Properties | It is formed in situ by the reaction between the components of the aqueous electrolyte and the zinc negative electrode. It is an organic-inorganic composite membrane that can conduct Zn2+; however, it has a loose structure and poor stability, making it difficult to block water molecules and prone to corrosion, hydrogen evolution and dendrite formation | Magnesium metal has extremely high reactivity. When it comes into contact with water or common solvents, it spontaneously forms a dense and inert passivation layer, preventing the conduction of Mg2+ ions and completely blocking the ion transport. It is not a functional SEI (solid electrolyte interface), and a stable interface capable of conducting ions can only be artificially constructed using special electrolytes |
| The core challenge of interface stability | 1. The hydrogen evolution reaction induced by free water in the aqueous electrolyte and the negative electrode corrosion[236] 2. Zinc dendrite growth caused by uneven deposition of Zn2+[237] 3. Positive electrode material dissolution and by-product deposition triggered by interface pH fluctuations 4. The narrow electrochemical window of traditional aqueous electrolyte 5. Poor interface contact of quasi-solid-state electrolyte | 1. In most conventional electrolytes, an insulating passivation layer tends to form on the surface of the magnesium anode[238] 2. At the positive electrode/ electrolyte interface, the energy barrier for Mg2+ desolvation is high, resulting in severe voltage lag 3. The interfacial side reactions between the high-pressure oxide positive electrode and the organic electrolyte are intense[239] 4. In solid-state magnesium-ion batteries, the Mg2+ conductivity at the solid-solid interface is low 5. During the cycling process, the interface structure of the conversion-type positive electrode deteriorates |
| The mainstream optimization strategies for interface stability | The core of water control activity, stable SEI formation, inhibition of dendrite growth, and reduction of water-related side reactions are achieved through electrolyte engineering (such as high-concentration salt in water), artificial SEI construction, modification of the negative electrode structure, and regulation and optimization of positive electrode CEI to improve interface stability[240-242] | The core of this approach is to avoid passivation, weaken ionic association, and construct a Mg2+ conductive interface. It involves the design of non-polar electrolytes, control of solvation shells, artificial interface modification, and negative electrode alloying and host design to improve interface performance[243,244] |
In terms of the logic of differentiated design, the core differences between the two systems stem from the choice of the electrolyte matrix, which in turn give rise to completely different core challenges and optimization priorities. For ZIBs, the aqueous electrolyte matrix offers inherent advantages such as high ionic conductivity, high safety, and low cost. However, the core contradiction lies in the water-induced side reactions (hydrogen evolution, negative electrode corrosion, and positive electrode dissolution) and the narrow electrochemical window. Therefore, the core of optimizing the electrolyte for ZIBs lies in maintaining the advantages of the aqueous system while reducing the activity of free water, and at the same time regulating the deposition behavior of Zn2+ to inhibit dendrite growth. For MIBs, the non-aqueous organic electrolyte matrix has overcome the voltage limit of water, laying the foundation for high energy density. However, the core issue is the magnesium anode passivation caused by electrolyte decomposition, as well as the sluggish transport kinetics of Mg2+ at the interface. Therefore, the core of optimizing the electrolyte for magnesium-ion batteries is to enhance the compatibility between the electrolyte and the magnesium anode, avoid the formation of insulating passivation layers, and simultaneously reduce the de-solvation energy barrier of Mg2+ at the positive electrode interface.
CONCLUSION
With countries worldwide striving for carbon neutrality and expanding the share of renewable energy, advanced energy storage has emerged as a core research priority. Despite their market dominance, LIBs suffer from uneven lithium reserves, volatile material prices and intrinsic safety defects, restricting their extensive grid-scale deployment. Under such constraints, ZIBs and MIBs stand out as competitive multivalent-ion alternatives. Offering inherent advantages such as abundant raw material reserves, low cost, high theoretical capacity, and excellent safety, these technologies have emerged as highly competitive alternatives to LIBs. Driven by recent breakthroughs in high-performance electrode materials, stable electrolytes, and interfacial engineering, the industrialization of ZIBs and MIBs is rapidly accelerating. To facilitate this trajectory, this review systematically summarizes the current research progress of ZIBs and MIBs, focusing on their electrochemical reaction mechanisms, electrode materials, electrolytes, and electrode/electrolyte modification strategies. Furthermore, we clarify existing technical bottlenecks and explore future development pathways, thereby providing a comprehensive reference for the practical implementation of these advanced battery technologies.
DECLARATIONS
Authors’ contribution
Contributed equally to this work, data sourcing, collection, conceptulization, and original draft writing: Liu, T.; Yin, J.
Data sourcing, collection: Wang, Q.; Yu, L.; Xu, J.
Editing and supervision: Tai, X.; Wang, S.; Wang, X.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (U21A20290, 22406028), and Maoming Science and Technology Special Fund Project (MMSZX2025001).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication.
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Anwar, H.; Waheed, R.; Aziz, G. Importance of FinTech and green finance to achieve the carbon neutrality targets: a study of australian perspective. Environ. Res. Commun. 2024, 6, 115007.
2. Zhang, W.; He, L.; Li, J.; et al. Configurational entropy-tailored NASICON cathode redox chemistry for capacity-dense and ultralong cyclability. Energy. Environ. Sci. 2025, 18, 7278-90.
3. Li, G.; Jin, Y.; Akram, M.; Chen, X. Research and current status of the solar photovoltaic water pumping system - a review. Renew. Sustain. Energy. Rev. 2017, 79, 440-58.
4. Kelly-richards, S.; Silber-coats, N.; Crootof, A.; Tecklin, D.; Bauer, C. Governing the transition to renewable energy: a review of impacts and policy issues in the small hydropower boom. Energy. Policy. 2017, 101, 251-64.
5. Liu, C.; Wu, X.; Wang, B. Performance modulation of energy storage devices: a case of Ni-Co-S electrode materials. Chem. Eng. J. 2020, 392, 123651.
6. Al-hallaj, S.; Wilk, G.; Crabtree, G.; Eberhard, M. Overview of distributed energy storage for demand charge reduction. MRS. Energy. Sustain. 2018, 5, 16.
7. Liu, D. H.; Bai, Z.; Li, M.; et al. Developing high safety Li-metal anodes for future high-energy Li-metal batteries: strategies and perspectives. Chem. Soc. Rev. 2020, 49, 5407-45.
8. Zheng, J.; Kim, M. S.; Tu, Z.; Choudhury, S.; Tang, T.; Archer, L. A. Regulating electrodeposition morphology of lithium: towards commercially relevant secondary Li metal batteries. Chem. Soc. Rev. 2020, 49, 2701-50.
9. Cano, Z. P.; Banham, D.; Ye, S.; et al. Batteries and fuel cells for emerging electric vehicle markets. Nat. Energy. 2018, 3, 279-89.
10. Chao, D.; Zhou, W.; Xie, F.; et al. Roadmap for advanced aqueous batteries: from design of materials to applications. Sci. Adv. 2020, 6, eaba4098.
11. Zubi, G.; Dufo-lópez, R.; Carvalho, M.; Pasaoglu, G. The lithium-ion battery: state of the art and future perspectives. Renew. Sustain. Energy. Rev. 2018, 89, 292-308.
12. Dubal, D. P.; Chodankar, N. R.; Kim, D. H.; Gomez-Romero, P. Towards flexible solid-state supercapacitors for smart and wearable electronics. Chem. Soc. Rev. 2018, 47, 2065-129.
13. Shi, Y.; Chen, Y.; Shi, L.; et al. An overview and future perspectives of rechargeable zinc batteries. Small 2020, 16, 2000730.
14. Tang, B.; Shan, L.; Liang, S.; Zhou, J. Issues and opportunities facing aqueous zinc-ion batteries. Energy. Environ. Sci. 2019, 12, 3288-304.
15. Arya, A.; Bag, S. Overcoming the challenges in aqueous zinc metal batteries: underlying issues and mitigation strategies. Chem. Asian. J. 2025, 20, e70004.
16. Khan, H.; Zhao, C.; Khan, K.; et al. Challenges and design strategies for stable zinc anodes in rechargeable zinc batteries. Small 2025, 21, 2504170.
17. Wang, X.; Zhang, Z.; Xi, B.; et al. Advances and perspectives of cathode storage chemistry in aqueous zinc-ion batteries. ACS. Nano. 2021, 15, 9244-72.
18. Fu, J.; Liang, R.; Liu, G.; et al. Recent progress in electrically rechargeable zinc-air batteries. Adv. Mater. 2019, 31, 1805230.
19. Liu, Y.; Chen, J.; Li, W.; et al. Aqueous Zn-CO2 batteries: a route towards sustainable energy storage. Ind. Chem. Mater. 2024, 2, 514-32.
20. Zhao, Y.; Zheng, M. Battery management system for zinc-based flow batteries: a review. Renew. Sustain. Energy. Rev. 2025, 215, 115604.
21. Yuan, Z.; Yin, Y.; Xie, C.; Zhang, H.; Yao, Y.; Li, X. Advanced materials for zinc-based flow battery: development and challenge. Adv. Mater. 2019, 31, 1902025.
22. Xiao, X.; Zheng, Z.; Zhong, X.; et al. Rational design of flexible Zn-based batteries for wearable electronic devices. ACS. Nano. 2023, 17, 1764-802.
23. Li, Y.; Fu, J.; Zhong, C.; et al. Recent advances in flexible zinc-based rechargeable batteries. Adv. Energy. Mater. 2019, 9, 1802605.
24. Zhu, Y.; Guan, P.; Zhu, R.; et al. Recent advances in flexible alkaline zinc-based batteries: materials, structures, and perspectives. J. Energy. Chem. 2023, 87, 61-88.
25. Innocenti, A.; Bresser, D.; Garche, J.; Passerini, S. A critical discussion of the current availability of lithium and zinc for use in batteries. Nat. Commun. 2024, 15, 4068.
27. Lu, H.; Zhang, D.; Zhu, Z.; et al. Three-in-one zinc anodes created by a large-scale two-step method achieving excellent long-term cyclic reversibility and thin electrode integrity. Adv. Sci. 2024, 11, e2401575.
28. Pang, Q.; Yu, X.; Zhang, S.; et al. High-capacity and long-lifespan aqueous LiV3O8/Zn battery using Zn/Li hybrid electrolyte. Nanomaterials 2021, 11, 1429.
29. Xue, T.; Mu, Y.; Wei, X.; et al. From fundamentals to practice: electrolyte strategies for zinc-ion batteries in extreme temperature. Carbon. Neutraliz. 2025, 4, e183.
30. Zhang, W.; Chen, J.; Guan, C.; et al. Harnessing dual hydrogen bonding and lewis acid-base interactions for bio-inspired symmetry-breaking electrolytes in aqueous zinc-ion batteries. Angew. Chem. Int. Ed. 2025, 64, e202516282.
31. Song, J.; Zhang, Q.; Yang, G.; et al. Biomimetic design for zinc-based energy storage devices: principles, challenges and opportunities. Chem. Soc. Rev. 2025, 54, 8071-135.
32. Alawi, M. J.; Gamal, H.; Rashad, M.; Alziyadi, M. O.; Shalaby, M. Zinc-ion batteries: drawbacks, opportunities, and optimization performance for sustainable energy storage. J. Alloys. Compd. 2025, 1012, 178455.
33. Miao, W.; Peng, H.; Cui, S.; et al. Fine nanostructure design of metal chalcogenide conversion-based cathode materials for rechargeable magnesium batteries. iScience 2024, 27, 109811.
34. Yan, X.; Luo, S.; Liu, Q.; et al. Insertion-type cathode materials in magnesium ion batteries: research progress and application potential. Mater. Today. Energy. 2025, 53, 102001.
35. Pang, Y.; Zhu, Y.; Fang, F.; Sun, D.; Zheng, S. Advances in solid Mg-ion electrolytes for solid-state Mg batteries. J. Mater. Sci. Technol. 2023, 161, 136-49.
36. Herodotou, P.; Hao, R.; Georghiou, G.; Yagi, S. Rechargeable magnesium batteries: system-level opportunities and challenges for battery energy storage applications. Electrochim. Acta. 2026, 555, 148315.
37. Yoo, H. D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg rechargeable batteries: an on-going challenge. Energy. Environ. Sci. 2013, 6, 2265.
38. Song, J.; Sahadeo, E.; Noked, M.; Lee, S. B. Mapping the challenges of magnesium battery. J. Phys. Chem. Lett. 2016, 7, 1736-49.
39. Zhang, Z.; Dong, S.; Cui, Z.; Du, A.; Li, G.; Cui, G. Rechargeable magnesium batteries using conversion-type cathodes: a perspective and minireview. Small. Methods. 2018, 2, 1800020.
40. Tang, H.; Peng, Z.; Wu, L.; et al. Vanadium-based cathode materials for rechargeable multivalent batteries: challenges and opportunities. Electrochem. Energy. Rev. 2018, 1, 169-99.
41. Xu, H.; He, X.; Li, Y.; et al. Reassessing electrolyte design for non-aqueous magnesium batteries: atomistic structures and performance optimization. Adv. Mater. 2026, 38, e14224.
42. Upreti, B. B.; Kamboj, N.; Dey, R. S. Advancing zinc anodes: strategies for enhanced performance in aqueous zinc-ion batteries. Small 2025, 21, 2408138.
43. Wang, S.; Liu, L.; Wu, J. Research status and optimization methods of zinc ion battery. MATEC. Web. Conf. 2023, 382, 01015.
44. Guo, Z.; Zhao, S.; Li, T.; Su, D.; Guo, S.; Wang, G. Recent advances in rechargeable magnesium-based batteries for high-efficiency energy storage. Adv. Energy. Mater. 2020, 10, 1903591.
45. Lei, X.; Liang, X.; Yang, R.; et al. Rational design strategy of novel energy storage systems: toward high-performance rechargeable magnesium batteries. Small 2022, 18, e2200418.
46. Xiao, D.; Lv, X.; Fan, J.; Li, Q.; Chen, Z. Zn-based batteries for energy storage. Energy. Mater. 2023, 3, 300007.
47. Li, Y.; Zhao, C.; Wu, X.; Bando, Y. NH4V4O10 cathode materials regulated by Co metal-organic frameworks for highly stable zinc battery at wide temperatures. Chem. Eng. J. 2025, 525, 169945.
48. Sun, S.; Wang, B.; Huang, K. Quantifying electrokinetics of NaCa0.6V6O16·3H2O cathode in aqueous zinc-ion batteries with ZnSO4 electrolyte. J. Mater. Chem. A. 2025, 13, 27189-99.
49. Xu, Y.; Cai, P.; Chen, K.; et al. High-voltage rechargeable alkali-acid Zn-PbO2 hybrid battery. Angew. Chem. Int. Ed. 2020, 59, 23593-7.
50. Ma, L.; Li, N.; Long, C.; et al. Achieving both high voltage and high capacity in aqueous zinc-ion battery for record high energy density. Adv. Funct. Mater. 2019, 29, 1906142.
51. Wang, S.; Yuan, Z.; Zhang, X.; et al. Non-metal ion Co-insertion chemistry in aqueous Zn/MnO2 batteries. Angew. Chem. Int. Ed. 2021, 60, 7056-60.
52. Cui, M.; Zhu, Y.; Lei, H.; et al. Anion-cation competition chemistry for comprehensive high-performance Prussian blue analogs cathodes. Angew. Chem. Int. Ed. 2024, 63, e202405428.
53. Ma, L.; Chen, S.; Long, C.; et al. Achieving high-voltage and high-capacity aqueous rechargeable zinc ion battery by incorporating two-species redox reaction. Adv. Energy. Mater. 2019, 9, 1902446.
54. Gu, X.; Wang, J.; Zhao, X.; et al. Engineered nitrogen doping on VO2(B) enables fast and reversible zinc-ion storage capability for aqueous zinc-ion batteries. J. Energy. Chem. 2023, 85, 30-8.
55. Muthuraj, D.; Mitra, S. Reversible Mg insertion into chevrel phase Mo6S8 cathode: Preparation, electrochemistry and X-ray photoelectron spectroscopy study. Mater. Res. Bull. 2018, 101, 167-74.
56. Li, C.; Wu, W.; Liu, Y.; et al. Facilitating Mg2+ diffusion in high potential LixV2(PO4)3 cathode material with a co-insertion strategy for rechargeable Mg-ion batteries. J. Power. Sources. 2022, 520, 230853.
57. Sun, C.; Wang, H.; Yang, F.; et al. Layered buserite Mg-Mn oxide cathode for aqueous rechargeable Mg-ion battery. J. Magnes. Alloys. 2023, 11, 840-50.
58. Wang, J.; Zhang, Y.; Liu, G.; et al. Improvements in the magnesium ion transport properties of graphene/CNT-wrapped TiO2-B nanoflowers by nickel doping. Small 2024, 20, 2304969.
59. Cai, R.; Qin, H.; Yu, X.; et al. Kirkendall effect-assisted synthesis of hollow MoS2 nanospheres with interlayer expansion for improved magnesium diffusion kinetics and durability. J. Mater. Chem. A. 2025, 13, 2574-82.
60. Wang, L.; Welborn, S. S.; Kumar, H.; et al. High-rate and long cycle-life alloy-type magnesium-ion battery anode enabled through (de)magnesiation-induced near-room-temperature solid-liquid phase transformation. Adv. Energy. Mater. 2019, 9, 1902086.
61. Zhao, M.; Ren, C. E.; Alhabeb, M.; Anasori, B.; Barsoum, M. W.; Gogotsi, Y. Magnesium-ion storage capability of MXenes. ACS. Appl. Energy. Mater. 2019, 2, 1572-8.
62. Volta, A. On the electricity excited by the mere contact of conducting substances of different kinds. In a letter from Mr. Alexander Volta, F. R. S. Professor of Natural Philosophy in the University of Pavia, to the Rt. Hon. Sir Joseph Banks, Bart. K. B. P. R. S. Proc. R. Soc. 1832, 1, 27-9.
63. Hao, J.; Li, X.; Zeng, X.; Li, D.; Mao, J.; Guo, Z. Deeply understanding the Zn anode behaviour and corresponding improvement strategies in different aqueous Zn-based batteries. Energy. Environ. Sci. 2020, 13, 3917-49.
64. Zhang, P.; Cai, M.; Wei, Y.; et al. Photo-assisted rechargeable metal batteries: principles, progress, and perspectives. Adv. Sci. 2024, 11, e2402448.
65. Zhou, X.; Jiang, S.; Zhu, S.; et al. The progress of cathode materials in aqueous zinc-ion batteries. Nanotechnol. Rev. 2023, 12, 20230122.
66. Xu, C.; Li, B.; Du, H.; Kang, F. Energetic zinc ion chemistry: the rechargeable zinc ion battery. Angew. Chem. Int. Ed. 2012, 51, 933-5.
67. Zhao, D.; Liu, C.; Chen, T.; Li, M. Recent advances in aqueous zinc ion batteries: energy storage mechanisms, challenges, and optimization strategies. Batteries 2026, 12, 109.
68. Zhao, C.; Zhou, Y.; Liu, Y.; et al. Advancing zinc-manganese oxide batteries: mechanistic insights, anode engineering, and cathode regulation. Nanomaterials 2025, 15, 1439.
69. Sun, W.; Wang, F.; Hou, S.; et al. Zn/MnO2 battery chemistry with H+ and Zn2+ coinsertion. J. Am. Chem. Soc. 2017, 139, 9775-8.
70. Zhao, Q.; Chen, X.; Wang, Z.; et al. Unravelling H+/Zn2+ synergistic intercalation in a novel phase of manganese oxide for high-performance aqueous rechargeable battery. Small 2019, 15, 1904545.
71. Pan, H.; Shao, Y.; Yan, P.; et al. Reversible aqueous zinc/manganese oxide energy storage from conversion reactions. Nat. Energy. 2016, 1, 16039.
72. Liu, W.; Zhang, X.; Huang, Y.; et al. β-MnO2 with proton conversion mechanism in rechargeable zinc ion battery. J. Energy. Chem. 2021, 56, 365-73.
73. Wang, T.; Li, C.; Xie, X.; et al. Anode materials for aqueous zinc ion batteries: mechanisms, properties, and perspectives. ACS. Nano. 2020, 14, 16321-47.
74. Shen, X.; Wang, X.; Zhou, Y.; et al. Highly reversible aqueous zn-MnO2 battery by supplementing Mn2+-mediated MnO2 deposition and dissolution. Adv. Funct. Mater. 2021, 31, 2101579.
75. Kim, S. H.; Oh, S. M. Degradation mechanism of layered MnO2 cathodes in Zn/ZnSO4/MnO2 rechargeable cells. J. Power. Sources. 1998, 72, 150-8.
76. Wu, T.; Lin, Y.; Althouse, Z. D.; Liu, N. Dissolution-redeposition mechanism of the MnO2 cathode in aqueous zinc-ion batteries. ACS. Appl. Energy. Mater. 2021, 4, 12267-74.
77. Chen, H.; Dai, C.; Xiao, F.; et al. Reunderstanding the reaction mechanism of aqueous Zn-Mn batteries with sulfate electrolytes: role of the zinc sulfate hydroxide. Adv. Mater. 2022, 34, 2109092.
78. Luan, J.; Yuan, H.; Liu, J.; Zhong, C. Recent advances on charge storage mechanisms and optimization strategies of Mn-based cathode in zinc-manganese oxides batteries. Energy. Storage. Mater. 2024, 66, 103206.
79. Yang, J.; Tian, H.; Wang, K.; Chen, J. 10 years of frontiers in materials: interface engineering for aqueous zinc-ion batteries. Front. Mater. 2024, 11, 1376865.
80. Li, Q.; Wang, C.; Zhu, Y.; et al. Unlocking the critical role of Mg doping in α-MnO2 cathode for aqueous zinc ion batteries. Chem. Eng. J. 2024, 485, 150077.
81. Chacón-borrero, J.; Chang, X.; Min, Z.; et al. Boosting high-loading zinc-ion battery performance: Zn-Doped δ-MnO2 cathodes to promote Zn2+ storage. Energy. Storage. Mater. 2025, 81, 104486.
82. Yang, S.; Li, F.; Fu, P.; et al. Room temperature synthesis of the Co-doped δ-MnO2 cathode for high-performance zinc-ion batteries. J. Power. Sources. 2024, 611, 234767.
83. Tian, M.; Zhu, C.; Luo, K. Hybrid organic-inorganic modification on tunnel-structured α-MnO2 for high-performance aqueous zinc-ion battery. Chin. Chem. Lett. 2026, 37, 110702.
84. Han, R.; Pan, Y.; Du, C.; et al. Eu doping β-MnO2 as cathode materials for high specific capacity aqueous zinc ion batteries. J. Energy. Storage. 2024, 80, 110250.
85. Paik, S.; Choi, I.; Lee, S.; Nam, K. W. Chelating effects of polyphenolic biomolecules to improve β-MnO2 cathode performance for aqueous rechargeable zinc-ion batteries. ACS. Appl. Mater. Interfaces. 2024, 16, 50775-84.
86. Li, S.; Zhao, M.; Zhang, D.; Wu, X. High-capacity aqueous Zn/MnO2 batteries: a clue of K ion preintercalation. Cryst. Growth. Des. 2023, 23, 8156-62.
87. Du, D.; Huang, C.; Liu, J.; et al. Amino-functionalized carbon nanotubes stimulating γ-MnO2 to achieve high-performance zinc-ion batteries. Electrochim. Acta. 2023, 456, 142461.
88. Gan, Y.; Shi, P.; Pan, J.; et al. High valence tungsten driven high stability phase and conductivity γ-MnO2 cathodes for durable aqueous zinc ion batteries. J. Energy. Storage. 2025, 127, 117176.
89. Paudel, N.; Ale Magar, B.; Acharya, K.; Lambert, T. N.; Vasiliev, I. Influence of defects and surfaces on the electrochemical performance of MnO2 cathodes in rechargeable alkaline Zn/MnO2 batteries: a first-principles study. ACS. Appl. Energy. Mater. 2024, 7, 2767-78.
90. He, Q.; Hu, T.; Wu, Q.; et al. Tunnel-oriented VO2(B) cathode for high-rate aqueous zinc-ion batteries. Adv. Mater. 2024, 36, 2400888.
91. Li, X.; Hu, J.; Wang, B.; et al. Interface coupling engineering of nano flower-like porous carbon with V2O5 for enhancing rapid transport of zinc ions in aqueous zinc-vanadium batteries. Carbon 2025, 233, 119917.
92. Wen, H.; Kong, A.; Shen, Y.; Fan, Y.; Liu, J.; Chen, L. Acid etching V2O5 nanowires cathode for high-performance zinc-ion full batteries. Chem. Eng. J. 2025, 511, 162148.
93. Wu, M.; Shi, C.; Yang, J.; et al. The LiV3O8 superlattice cathode with optimized zinc ion insertion chemistry for high mass-loading aqueous zinc-ion batteries. Adv. Mater. 2024, 36, 2310434.
94. Wang, L.; Zhao, M.; Zhang, X.; et al. Novel high-voltage cathode for aqueous zinc ion batteries: porous K0.5VOPO4·1.5H2O with reversible solid-solution intercalation and conversion storage mechanism. J. Energy. Chem. 2024, 93, 71-8.
95. Zampardi, G.; La Mantia, F. Prussian blue analogues as aqueous Zn-ion batteries electrodes: current challenges and future perspectives. Curr. Opin. Electrochem. 2020, 21, 84-92.
96. Syed, W. A.; Kakarla, A. K.; Bandi, H.; Shanthappa, R.; Yu, J. S. Copper substituted manganese Prussian blue analogue composite nanostructures for efficient aqueous zinc-ion batteries. J. Energy. Storage. 2024, 99, 113325.
97. Yang, G.; Liang, Z.; Li, Q.; Li, Y.; Tian, F.; Wang, C. Epitaxial core-shell MnFe Prussian blue cathode for highly stable aqueous zinc batteries. ACS. Energy. Lett. 2023, 8, 4085-95.
98. Sun, Y.; Xu, Z.; Xu, X.; et al. Low-cost and long-life Zn/Prussian blue battery using a water-in-ethanol electrolyte with a normal salt concentration. Energy. Storage. Mater. 2022, 48, 192-204.
99. Xie, M.; Zhang, X.; Wang, R.; et al. Mn-O bond engineering mitigating Jahn-Teller effects of manganese oxide for aqueous zinc-ion battery applications. Chem. Eng. J. 2024, 494, 152908.
100. Peng, G.; Shao, F.; Bian, Y.; et al. Suppressing Jahn-Teller effect in manganese-based oxides for aqueous zinc-ion batteries through advanced composite material engineering. J. Energy. Storage. 2025, 134, 118216.
101. Jun, S. J.; Lee, J.; Ryu, M.; et al. Electrode interface modulation using 1,3-propane sultone functionalized additives for high-performance Zn-MnO2 aqueous batteries. Chem. Eng. J. 2024, 497, 154394.
102. Wang, W.; Yang, C.; Chi, X.; Liu, J.; Wen, B.; Liu, Y. Ultralow-water-activity electrolyte endows vanadium-based zinc-ion batteries with durable lifespan exceeding 30000 cycles. Energy. Storage. Mater. 2022, 53, 774-82.
103. Xiong, Y.; Li, Q.; Luo, K.; et al. Low cost carboxymethyl cellulose additive toward stable zinc anodes in aqueous zinc ion battery. J. Energy. Storage. 2023, 68, 107655.
104. Park, S.; An, G. Improvement of structural stability of cathode by manganese additive in electrolyte for zinc-ion batteries. Int. J. Energy. Res. 2022, 46, 8464-70.
105. Chazalviel, J. Electrochemical aspects of the generation of ramified metallic electrodeposits. Phys. Rev. A. 1990, 42, 7355-67.
106. Brissot, C.; Rosso, M.; Chazalviel, J.; Lascaud, S. Dendritic growth mechanisms in lithium/polymer cells. J. Power. Sources. 1999, 81-82, 925-9.
107. Hu, X.; Lin, Z.; Wang, S.; et al. Highly crystalline flower-like covalent-organic frameworks enable highly stable zinc metal anodes. ACS. Appl. Energy. Mater. 2022, 5, 3715-23.
108. Zong, Q.; Liu, X.; Zhang, Q.; et al. Interfacial gradient engineering synergized with self-adaptive cathodic defense for durable Zn-ion batteries. Energy. Environ. Sci. 2025, 18, 8256-67.
109. Hyun, D.; Choi, J. C.; Sim, J. S.; et al. Scalable electrodeposition method to construct zincophilic 3D Cu nanosheet current collector for Zn metal anode. Chem. Eng. J. 2025, 508, 161071.
110. Zhang, Y.; Yang, X.; Hu, Y.; et al. Highly strengthened and toughened Zn-Li-Mn alloys as long-cycling life and dendrite-free Zn anode for aqueous zinc-ion batteries. Small 2022, 18, e2200787.
111. Jiang, L.; Chai, Y.; Ji, D.; et al. Construction of an artificial zinc alloy layer toward stable zinc-metal anode. Green. Energy. Environ. 2025, 10, 382-9.
112. Jeon, H. G.; Heo, S.; Song, J.; Kim, C. A near-single-ion conducting protective layer for dendrite-free zinc metal anodes. Adv. Energy. Mater. 2025, 15, 2500155.
113. Lv, Y.; Xiao, Y.; Ma, L.; Zhi, C.; Chen, S. Recent advances in electrolytes for “beyond aqueous” zinc-ion batteries. Adv. Mater. 2022, 34, 2106409.
114. Meng, Q.; Yan, T.; Wang, Y.; Lu, X.; Zhou, H.; Dong, S. Critical design strategy of electrolyte engineering toward aqueous zinc-ion battery. Chem. Eng. J. 2024, 497, 154541.
115. Song, P.; Wang, Z.; Liu, W.; et al. A bifunctional biomass-derived additive for constructing a hybrid SEI layer to enhance the cycling stability of aqueous zinc-ion batteries under high current densities. Inorg. Chem. Front. 2026, 13, 3772-85.
116. Chen, T. Y.; Lin, T. J.; Vedhanarayanan, B.; Shen, H. H.; Lin, T. W. Optimization of acetamide based deep eutectic solvents with dual cations for high performance and low temperature-tolerant aqueous zinc ion batteries via tuning the ratio of co-solvents. J. Colloid. Interface. Sci. 2023, 629, 166-78.
117. Yu, Q.; Shen, T.; Ke, S. W.; et al. Ni-Bis(dithiolene) coordination enhanced dual-functional covalent organic frameworks for both cathodic Zn2+ storage and anodic zinc deposition control in aqueous Zn-ion batteries. Angew. Chem. Int. Ed. 2025, 64, e202507352.
118. Li, P.; Ren, J.; Li, C.; et al. MOF-derived defect-rich CeO2 as ion-selective smart artificial SEI for dendrite-free Zn-ion battery. Chem. Eng. J. 2023, 451, 138769.
119. Chen, D.; Wang, H.; Ren, L.; et al. Zinc-ion conductive buffer polymer layer eliminating parasitic reactions of Zn anode in aqueous zinc-ion batteries. Sci. China. Mater. 2023, 66, 4605-14.
120. Zhang, Y.; Zheng, X.; Wang, N.; et al. Anode optimization strategies for aqueous zinc-ion batteries. Chem. Sci. 2022, 13, 14246-63.
121. Zhang, X.; Hu, J.; Fu, N.; et al. Comprehensive review on zinc-ion battery anode: challenges and strategies. InfoMat 2022, 4, e12306.
122. Verma, V.; Kumar, S.; Manalastas, W.; Srinivasan, M. Undesired reactions in aqueous rechargeable zinc ion batteries. ACS. Energy. Lett. 2021, 6, 1773-85.
123. Li, G.; Sun, L.; Zhang, S.; et al. Developing cathode materials for aqueous zinc ion batteries: challenges and practical prospects. Adv. Funct. Mater. 2024, 34, 2301291.
124. Sun, L.; Song, Z.; Deng, C.; et al. Electrolytes for aqueous Zn-ion batteries working in wide-temperature range: progress and perspective. Batteries 2023, 9, 386.
125. Kang, Y.; Zhang, F.; Li, H.; et al. Modulating the electrolyte inner solvation structure via low polarity co-solvent for low-temperature aqueous zinc-ion batteries. Energy. Environ. Mater. 2024, 7, e12707.
126. Dong, Y.; Chai, X.; Li, J.; et al. High-entropy electrolytes downsizing solvated clusters and enabling parallel ion transport for low-temperature aqueous zinc batteries. J. Am. Chem. Soc. 2025, 147, 35544-54.
127. Chen, Z.; Zhou, W.; Zhao, S.; Lou, X.; Chen, S. In-situ construction of solid electrolyte interphases with gradient zincophilicity for wide temperature zinc ion batteries. Adv. Energy. Mater. 2025, 15, 2404108.
128. Li, S.; Wang, S.; Zhu, J. Advanced dual-anionic electrolyte additive for high-performance aqueous zinc-ion batteries. Chem. Eng. J. 2025, 523, 168682.
129. Shi, Y.; Wang, R.; Bi, S.; Yang, M.; Liu, L.; Niu, Z. An anti-freezing hydrogel electrolyte for flexible zinc-ion batteries operating at -70 °C. Adv. Funct. Mater. 2023, 33, 2214546.
130. Samanta, P.; Ghosh, S.; Kolya, H.; Kang, C. W.; Murmu, N. C.; Kuila, T. Molecular crowded “Water-in-Salt” polymer gel electrolyte for an ultra-stable Zn-ion battery. ACS. Appl. Mater. Interfaces. 2022, 14, 1138-48.
131. Yu, Y.; Li, D.; Guo, Q.; et al. Anion-induced optimization of non-aqueous zinc-air battery performance: Insights into OTF- selection. J. Power. Sources. 2025, 626, 235764.
132. Yao, X.; Wang, Z.; Guo, J.; et al. Paschen-Back effect modulation of SO42- hydration in magnetized electrolyte toward dendrite-free Zn-ion batteries. Nat. Commun. 2025, 16, 5740.
133. Cui, M.; Yu, L.; Hu, J.; He, S.; Zhi, C.; Huang, Y. Tailored polymer-inorganic bilayer SEI with proton holder feature for aqueous Zn metal batteries. Angew. Chem. Int. Ed. 2025, 64, e202423531.
134. Xing, Z.; Xu, G.; Han, J.; et al. Facing the capacity fading of vanadium-based zinc-ion batteries. Trends. Chem. 2023, 5, 380-92.
135. Zhang, X.; Liu, Y.; Wang, S.; et al. Fundamentals and design strategies of electrolytes for high-temperature zinc-ion batteries. Energy. Storage. Mater. 2024, 70, 103471.
136. Shang, Y.; Tong, Z.; Kundu, D. Decoding the zinc depletion-mediated failure in aqueous zinc batteries: on limiting parameters and accurate assessment. ACS. Energy. Lett. 2024, 9, 3084-92.
137. Kim, Y.; Park, Y.; Kim, M.; Lee, J.; Kim, K. J.; Choi, J. W. Corrosion as the origin of limited lifetime of vanadium oxide-based aqueous zinc ion batteries. Nat. Commun. 2022, 13, 2371.
138. Chen, M.; Xie, S.; Zhao, X.; et al. Aqueous zinc-ion batteries at extreme temperature: mechanisms, challenges, and strategies. Energy. Storage. Mater. 2022, 51, 683-718.
139. Zhao, C.; Wu, M.; Lu, W.; et al. Electrochemical failure mechanism of δ-MnO2 in zinc ion batteries induced by irreversible layered to spinel phase transition. Small 2024, 20, 2401379.
140. Zhang, L.; Liu, Y. Aqueous zinc-chalcogen batteries: emerging conversion-type energy storage systems. Batteries 2023, 9, 62.
141. Chen, R.; Zhang, W.; Huang, Q.; et al. Trace amounts of triple-functional additives enable reversible aqueous zinc-ion batteries from a comprehensive perspective. NanoMicro. Lett. 2023, 15, 81.
142. Hong, L.; Guan, J.; Tan, Y.; et al. An effective descriptor for the screening of electrolyte additives toward the stabilization of Zn metal anodes. Energy. Environ. Sci. 2024, 17, 3157-67.
143. Aruchamy, K.; Ramasundaram, S.; Divya, S.; Chandran, M.; Yun, K.; Oh, T. H. Gel polymer electrolytes: advancing solid-state batteries for high-performance applications. Gels 2023, 9, 585.
144. Li, P.; Wang, Q. Novel Structural janus hydrogels for battery applications: structure design, properties, and prospects. Colloids. Interfaces. 2025, 9, 48.
145. Liu, S.; Zhang, R.; Wang, C.; et al. Zinc ion batteries: bridging the gap from academia to industry for grid-scale energy storage. Angew. Chem. Int. Ed. 2024, 63, e202400045.
146. Zhang, R.; Fan, Y.; Li, J.; et al. Constructing highly reversible zinc batteries under high depth of discharge & current density conditions via quaternary ammonium cations modulating electric field force and competitive solvation. J. Mater. Chem. A. 2025, 13, 26581-92.
147. Song, Y.; Chen, M.; Zhong, Z.; Liu, Z.; Liang, S.; Fang, G. Bilateral in-situ functionalization towards Ah-scale aqueous zinc metal batteries. Nat. Commun. 2025, 16, 3142.
148. Shi, S.; Zhou, D.; Jiang, Y.; et al. Lightweight Zn-philic 3D-Cu scaffold for customizable zinc ion batteries. Adv. Funct. Mater. 2024, 34, 2312664.
149. Zhao, Z.; Shang, F.; Liang, X.; et al. Enhancing interface kinetics in zinc powder anodes via β-cyclodextrin modification toward zinc ion batteries with low N/P ratios. Green. Chem. 2026, 28, 2056-65.
150. Han, R.; Jiang, T.; Wang, Z.; et al. Reconfiguring Zn2+ solvation network and interfacial chemistry of Zn metal anode with molecular engineered crown ether additive. Adv. Funct. Mater. 2025, 35, 2412255.
151. Aurbach, D.; Lu, Z.; Schechter, A.; et al. Prototype systems for rechargeable magnesium batteries. Nature 2000, 407, 724-7.
152. Mao, M.; Gao, T.; Hou, S.; Wang, C. A critical review of cathodes for rechargeable Mg batteries. Chem. Soc. Rev. 2018, 47, 8804-41.
153. Muldoon, J.; Bucur, C. B.; Gregory, T. Quest for nonaqueous multivalent secondary batteries: magnesium and beyond. Chem. Rev. 2014, 114, 11683-720.
154. Novák, P.; Imhof, R.; Haas, O. Magnesium insertion electrodes for rechargeable nonaqueous batteries - a competitive alternative to lithium? Electrochim. Acta. 1999, 45, 351-67.
155. Besenhard, J. O.; Winter, M. Advances in battery technology: rechargeable magnesium batteries and novel negative-electrode materials for lithium ion batteries. ChemPhysChem 2002, 3, 155-9.
156. Sun, R.; Pei, C.; Sheng, J.; et al. High-rate and long-life VS2 cathodes for hybrid magnesium-based battery. Energy. Storage. Mater. 2018, 12, 61-8.
157. Yang, R.; Yao, W.; Tang, B.; et al. Development and challenges of electrode materials for rechargeable Mg batteries. Energy. Storage. Mater. 2021, 42, 687-704.
158. Walter, M.; Kovalenko, M. V.; Kravchyk, K. V. Challenges and benefits of post-lithium-ion batteries. New. J. Chem. 2020, 44, 1677-83.
159. Zhang, J.; Cai, W.; Hu, F. X.; Yang, H.; Liu, B. Recent advances in single atom catalysts for the electrochemical carbon dioxide reduction reaction. Chem. Sci. 2021, 12, 6800-19.
160. Lin, D.; Liu, Y.; Cui, Y. Reviving the lithium metal anode for high-energy batteries. Nat. Nanotechnol. 2017, 12, 194-206.
161. Zhou, M.; Zhao, Y.; Yang, J.; Wang, J.; Nuli, Y. Polyanthraquinone with facile synthesis as a cathode material for rechargeable magnesium batteries. Energy. Fuels. 2024, 38, 19890-6.
162. Kim, H. S.; Arthur, T. S.; Allred, G. D.; et al. Structure and compatibility of a magnesium electrolyte with a sulphur cathode. Nat. Commun. 2011, 2, 427.
163. Gao, T.; Hou, S.; Wang, F.; et al. Reversible S0/MgSx redox chemistry in a MgTFSI2/MgCl2/DME electrolyte for rechargeable Mg/S batteries. Angew. Chem. Int. Ed. 2017, 56, 13526-30.
164. Zhao-Karger, Z.; Zhao, X.; Wang, D.; Diemant, T.; Behm, R. J.; Fichtner, M. Performance improvement of magnesium sulfur batteries with modified non-nucleophilic electrolytes. Adv. Energy. Mater. 2015, 5, 1401155.
165. Zhao-karger, Z.; Liu, R.; Dai, W.; et al. Toward highly reversible magnesium-sulfur batteries with efficient and practical Mg[B(hfip)4]2 electrolyte. ACS. Energy. Lett. 2018, 3, 2005-13.
166. Parambath, V. B.; Zhao-karger, Z.; Diemant, T.; et al. Investigation on the formation of Mg metal anode/electrolyte interfaces in Mg/S batteries with electrolyte additives. J. Mater. Chem. A. 2020, 8, 22998-3010.
167. Du, A.; Zhang, Z.; Qu, H.; et al. An efficient organic magnesium borate-based electrolyte with non-nucleophilic characteristics for magnesium-sulfur battery. Energy. Environ. Sci. 2017, 10, 2616-25.
168. Levi, E.; Lancry, E.; Mitelman, A.; Aurbach, D.; Isnard, O.; Djurado, D. Phase diagram of mg insertion into chevrel phases, MgxMo6T8 (T = S, Se). 2. The crystal structure of triclinic MgMo6Se8. Chem. Mater. 2006, 18, 3705-14.
169. Levi, M. D.; Lancry, E.; Gizbar, H.; et al. Kinetic and thermodynamic studies of Mg2+ and Li+ ion insertion into the Mo6S8 chevrel phase. J. Electrochem. Soc. 2004, 151, A1044.
170. Ling, C.; Suto, K. Thermodynamic origin of irreversible magnesium trapping in chevrel phase Mo6S8: importance of magnesium and vacancy ordering. Chem. Mater. 2017, 29, 3731-9.
171. Kang, G.; Hu, Q.; Li, S.; Bhoraskar, S. V.; Yoo, J. Synthesis of novel 1-dimensional structure from Mo6S8 chevrel phase of electrode for Mg batteries. Mater. Res. Express. 2022, 9, 085502.
172. Wang, D.; Du, X.; Chen, G.; et al. Cathode electrolyte interphase (CEI) endows Mo6S8 with fast interfacial magnesium-ion transfer kinetics. Angew. Chem. Int. Ed. 2023, 62, e202217709.
173. Gao, Y.; Xu, J.; Huang, K.; Lu, H.; Pang, Y.; Li, G. An overview of the current status and prospects of cathode materials based on transition metal sulfides for magnesium-ion batteries. CrystEngComm 2021, 23, 7546-64.
174. Zhang, H.; Ye, K.; Zhu, K.; et al. High-energy-density aqueous magnesium-ion battery based on a carbon-coated FeVO4 anode and a Mg-OMS-1 cathode. Chemistry 2017, 23, 17118-26.
175. Niu, J.; Gao, H.; Ma, W.; et al. Dual phase enhanced superior electrochemical performance of nanoporous bismuth-tin alloy anodes for magnesium-ion batteries. Energy. Storage. Mater. 2018, 14, 351-60.
176. Kotobuki, M.; Yan, B.; Lu, L. Recent progress on cathode materials for rechargeable magnesium batteries. Energy. Storage. Mater. 2023, 54, 227-53.
177. Yoo, H. D.; Liang, Y.; Dong, H.; et al. Fast kinetics of magnesium monochloride cations in interlayer-expanded titanium disulfide for magnesium rechargeable batteries. Nat. Commun. 2017, 8, 339.
178. Liang, Y.; Feng, R.; Yang, S.; Ma, H.; Liang, J.; Chen, J. Rechargeable Mg batteries with graphene-like MoS2 cathode and ultrasmall Mg nanoparticle anode. Adv. Mater. 2011, 23, 640-3.
179. Cheng, Y.; Shao, Y.; Raju, V.; et al. Molecular storage of Mg ions with vanadium oxide nanoclusters. Adv. Funct. Mater. 2016, 26, 3446-53.
180. Yin, J.; Pelliccione, C. J.; Lee, S. H.; Takeuchi, E. S.; Takeuchi, K. J.; Marschilok, A. C. Communication-sol-gel synthesized magnesium vanadium oxide, MgxV2O5·nH2O: the role of structural Mg2+ on battery performance. J. Electrochem. Soc. 2016, 163, A1941-3.
181. Zuo, C.; Tang, W.; Lan, B.; et al. Unexpected discovery of magnesium-vanadium spinel oxide containing extractable Mg2+ as a high-capacity cathode material for magnesium ion batteries. Chem. Eng. J. 2021, 405, 127005.
182. Pérez-Vicente, C.; Rubio, S.; Ruiz, R.; et al. Olivine-type MgMn0.5Zn0.5SiO4 cathode for Mg-batteries: experimental studies and first principles calculations. Small 2023, 19, e2206010.
183. Zhang, R.; Arthur, T. S.; Ling, C.; Mizuno, F. Manganese dioxides as rechargeable magnesium battery cathode; synthetic approach to understand magnesiation process. J. Power. Sources. 2015, 282, 630-8.
184. Nam, K. W.; Kim, S.; Lee, S.; et al. The high performance of crystal water containing manganese birnessite cathodes for magnesium batteries. Nano. Lett. 2015, 15, 4071-9.
185. Shen, J.; Zhang, Y.; Chen, D.; et al. A hollow CuS nanocube cathode for rechargeable Mg batteries: effect of the structure on the performance. J. Mater. Chem. A. 2019, 7, 21410-20.
186. Xiong, F.; Fan, Y.; Tan, S.; et al. Magnesium storage performance and mechanism of CuS cathode. Nano. Energy. 2018, 47, 210-6.
187. Wang, J.; Handoko, A. D.; Bai, Y.; et al. High-performance NiS2 hollow nanosphere cathodes in magnesium-ion batteries enabled by tunable redox chemistry. Nano. Lett. 2022, 22, 10184-91.
188. Roy, A.; Sotoudeh, M.; Dinda, S.; et al. Improving rechargeable magnesium batteries through dual cation co-intercalation strategy. Nat. Commun. 2024, 15, 492.
189. Sano, H.; Senoh, H.; Yao, M.; Sakaebe, H.; Kiyobayashi, T. Mg2+ storage in organic positive-electrode active material based on 2,5-dimethoxy-1,4-benzoquinone. Chem. Lett. 2012, 41, 1594-6.
190. Pan, B.; Huang, J.; Feng, Z.; et al. Polyanthraquinone-based organic cathode for high-performance rechargeable magnesium-ion batteries. Adv. Energy. Mater. 2016, 6, 1600140.
191. Wang, Z.; Diao, J.; Li, J.; Wang, J.; Tao, H.; Li, H. TiS2 nanosheets exfoliated by liquid phase ultrasound as cathode materials for magnesium ion batteries. Mater. Sci. Eng. B. 2026, 324, 119003.
192. Cheng, M.; Wang, Y.; Zhang, D.; et al. An efficient Hauser-base electrolyte for rechargeable magnesium batteries. J. Energy. Chem. 2023, 76, 1-10.
193. Liu, J.; Liu, J.; Zhou, T.; et al. High-performance magnesium/lithium hybrid-ion battery using NiS2 nanosheet-coated Cu2S cubes as a cathode material. ACS. Appl. Mater. Interfaces. 2025, 17, 38041-9.
194. Ye, Z.; Li, P.; Wei, W.; et al. In situ anchoring anion-rich and multi-cavity NiS2 nanoparticles on NCNTs for advanced magnesium-ion batteries. Adv. Sci. 2022, 9, e2200067.
195. Fichtner, M. Anodes for magnesium batteries: state-of-the-art and prospects. a viewpoint. J. Magnes. Alloys. 2025, 13, 4061-3.
196. Liu, F.; Wang, T.; Liu, X.; Fan, L. Challenges and recent progress on key materials for rechargeable magnesium batteries. Adv. Energy. Mater. 2021, 11, 2000787.
197. Chen, X.; Liu, X.; Le, Q.; Zhang, M.; Liu, M.; Atrens, A. A comprehensive review of the development of magnesium anodes for primary batteries. J. Mater. Chem. A. 2021, 9, 12367-99.
198. Wu, J.; Gao, G.; Wu, G.; et al. MgVPO 4 F as a one-dimensional Mg-ion conductor for Mg ion battery positive electrode: a first principles calculation. RSC. Adv. 2014, 4, 15014-7.
199. Wu, N.; Lyu, Y.; Xiao, R.; et al. A highly reversible, low-strain Mg-ion insertion anode material for rechargeable Mg-ion batteries. NPG. Asia. Mater. 2014, 6, e120-e120.
200. Zeng, J.; Yang, Y.; Li, C.; et al. Li3VO4: an insertion anode material for magnesium ion batteries with high specific capacity. Electrochim. Acta. 2017, 247, 265-70.
201. Tan, Y. H.; Yao, W. T.; Zhang, T.; et al. High voltage magnesium-ion battery enabled by nanocluster Mg3Bi2 alloy anode in noncorrosive electrolyte. ACS. Nano. 2018, 12, 5856-65.
202. Majid, A.; Ramzan, M.; Ahmad, S.; Alkhedher, M. Unraveling the potential of Al2CO bilayer as anode material in magnesium ion battery and unsuitability for lithium ion battery. J. Alloys. Compd. 2024, 981, 173697.
203. Tasawar, S.; Majid, A.; Ahmad, S.; Alkhedher, M.; Haider, S.; Alam, K. Uncovering the potential of layered InOCI as anode material in lithium, magnesium, and aluminum ion batteries: first-principles investigations. Battery. Energy. 2025, 4, e70013.
204. Zhang, R.; Cui, C.; Xiao, R.; et al. Interface regulation of Mg anode and redox couple conversion in cathode by copper for high-performance Mg-S battery. Chem. Eng. J. 2023, 451, 138663.
205. Wei, C.; Tan, L.; Zhang, Y.; et al. Highly reversible Mg metal anodes enabled by interfacial liquid metal engineering for high-energy Mg-S batteries. Energy. Storage. Mater. 2022, 48, 447-57.
206. Mizrahi, O.; Amir, N.; Pollak, E.; et al. Electrolyte solutions with a wide electrochemical window for rechargeable magnesium batteries. J. Electrochem. Soc. 2008, 155, A103.
207. Zhang, J.; Guan, X.; Lv, R.; Wang, D.; Liu, P.; Luo, J. Rechargeable Mg metal batteries enabled by a protection layer formed in vivo. Energy. Storage. Mater. 2020, 26, 408-13.
208. Li, Z.; Diemant, T.; Meng, Z.; et al. Establishing a stable anode-electrolyte interface in Mg batteries by electrolyte additive. ACS. Appl. Mater. Interfaces. 2021, 13, 33123-32.
209. Gregory, T. D.; Hoffman, R. J.; Winterton, R. C. Nonaqueous electrochemistry of magnesium: applications to energy storage. J. Electrochem. Soc. 1990, 137, 775-80.
210. Muldoon, J.; Bucur, C. B.; Oliver, A. G.; et al. Electrolyte roadblocks to a magnesium rechargeable battery. Energy. Environ. Sci. 2012, 5, 5941.
211. Zhang, Z.; Cui, Z.; Qiao, L.; et al. Novel design concepts of efficient Mg-ion electrolytes toward high-performance magnesium-selenium and magnesium-sulfur batteries. Adv. Energy. Mater. 2017, 7, 1602055.
212. Hassan, H. K.; Farkas, A.; Varzi, A.; Jacob, T. Mixed metal-organic frameworks as efficient semi-solid electrolytes for magnesium-ion batteries. Batteries. Supercaps. 2022, 5, e202200260.
213. Wang, J.; Zhao, W.; Dou, H.; et al. Electrostatic shielding guides lateral deposition for stable interphase toward reversible magnesium metal anodes. ACS. Appl. Mater. Interfaces. 2020, 12, 19601-6.
214. Pan, M.; Sun, Y.; Zhao, Y.; et al. Chelating anion-mediated solvation structures for rechargeable magnesium batteries. Angew. Chem. Int. Ed. 2026, 65, e6066611.
215. Fan, H.; Zhang, X.; Xiao, J.; et al. Tailoring electrode-electrolyte interface using an electron-deficient borate-based additive in MgTFSI2-MgCl2/DME electrolyte for rechargeable magnesium batteries. Energy. Environ. Mater. 2024, 7, e12792.
216. Zhang, R.; Cui, C.; Li, R.; et al. An artificial interphase enables the use of Mg(TFSI)2-based electrolytes in magnesium metal batteries. Chem. Eng. J. 2021, 426, 130751.
217. He, Y.; Li, Q.; Yang, L.; Yang, C.; Xu, D. Electrochemical-conditioning-free and water-resistant hybrid AlCl3/MgCl2/Mg(TFSI)2 electrolytes for rechargeable magnesium batteries. Angew. Chem. Int. Ed. 2019, 58, 7615-9.
218. Guzmán-torres, J.; González-juárez, E.; de la Luz Hernández-Nieto, M.; Espinosa-Roa, A.; Sánchez, E. M. Solid polymer electrolyte based on PEO/PVDF/Mg(ClO4)2-[EMIM][ESO4] system for rechargeable magnesium ion batteries. Ionics 2023, 29, 2341-9.
219. Zhao-Karger, Z.; Fichtner, M. Beyond intercalation chemistry for rechargeable Mg batteries: a short review and perspective. Front. Chem. 2018, 6, 656.
220. Itani, K.; De Bernardinis, A. Review on new-generation batteries technologies: trends and future directions. Energies 2023, 16, 7530.
221. Robba, A.; Tchernychova, E.; Bitenc, J.; Randon-vitanova, A.; Dominko, R. Magnesium insertion and related structural changes in spinel-type manganese oxides. Crystals 2021, 11, 984.
222. Bieker, G.; Küpers, V.; Kolek, M.; Winter, M. Intrinsic differences and realistic perspectives of lithium-sulfur and magnesium-sulfur batteries. Commun. Mater. 2021, 2, 143.
223. Ye, H.; Li, Y. Room-temperature metal-sulfur batteries: what can we learn from lithium-sulfur ? InfoMat 2022, 4, e12291.
224. Luo, T.; Wang, Y.; Elander, B.; et al. Polysulfides in magnesium-sulfur batteries. Adv. Mater. 2024, 36, e2306239.
225. Johnson, I. D.; Nolis, G.; Yin, L.; et al. Enhanced charge storage of nanometric ζ-V2O5 in Mg electrolytes. Nanoscale 2020, 12, 22150-60.
226. Mohammad, I.; Blondeau, L.; Leroy, J.; Khodja, H.; Gauthier, M. Influence of electrolyte on the electrode/electrolyte interface formation on InSb electrode in Mg-ion batteries. Molecules 2021, 26, 5721.
227. Bianchini, M.; Lacivita, V.; Seo, D.; Kim, H. Advances and challenges in multiscale characterizations and analyses for battery materials. J. Mater. Res. 2022, 37, 3113-29.
228. Cheng, W.; Zhao, M.; Lai, Y.; et al. Recent advances in battery characterization using in situ XAFS, SAXS, XRD, and their combining techniques: from single scale to multiscale structure detection. Exploration 2024, 4, 20230056.
229. Wang, J.; Liu, R.; Shen, Z.; Tian, J.; Wen, R. Recent progress in the application of in situ atomic force microscopy for metal anode processes in energy storage batteries. Chem. Phys. Rev. 2023, 4, 031308.
230. Siczek, K. Life-related hazards of materials applied to Mg-S batteries. Energies 2022, 15, 1543.
231. Li, Y.; Feng, X.; Lieu, W. Y.; et al. MXene-based anode-free magnesium metal battery. Adv. Funct. Mater. 2023, 33, 2303067.
232. Guo, W.; Bai, X.; Cong, Z.; et al. Suppressing the exacerbated hydrogen evolution of porous Zn anode with an artificial solid-electrolyte interphase layer. ACS. Appl. Mater. Interfaces. 2022, 14, 41988-96.
233. Chen, F.; Gao, Y.; Hao, Q.; Chen, X.; Sun, X.; Li, N. A 2.4 V aqueous zinc-ion battery enabled by the photoelectrochemical effect of a modified BiOI photocathode: shattering the shackle of the electrochemical window of an aqueous electrolyte. ACS. Nano. 2024, 18, 6413-23.
234. Sun, Y.; Zou, Q.; Wang, W.; Lu, Y. Non-passivating anion adsorption enables reversible magnesium redox in simple non-nucleophilic electrolytes. ACS. Energy. Lett. 2021, 6, 3607-13.
235. Sun, J.; Zou, Y.; Gao, S.; Shao, L.; Chen, C. Robust strategy of quasi-solid-state electrolytes to boost the stability and compatibility of Mg ion batteries. ACS. Appl. Mater. Interfaces. 2020, 12, 54711-9.
236. Huy, V. P.; Hieu, L. T.; Hur, J. Zn metal anodes for Zn-ion batteries in mild aqueous electrolytes: challenges and strategies. Nanomaterials 2021, 11, 2746.
237. Li, W.; Wang, S.; Hou, X.; et al. Enhancing ion dynamics and ion-enrichment through electronegative azolate framework for zinc anode stabilization. Chem. Eng. J. 2025, 520, 166235.
238. Liu, F.; Cao, G.; Ban, J.; et al. Recent advances based on Mg anodes and their interfacial modulation in Mg batteries. J. Magnes. Alloys. 2022, 10, 2699-716.
239. Liu, C.; Li, S.; Li, L.; et al. Li3InCl6 electrolyte with high voltage compatibility for Ni-rich layered oxide cathodes-based all-solid-state batteries. Electrochim. Acta. 2025, 535, 146568.
240. Wang, Z.; Ji, T.; Zhang, Q.; et al. Biodegradable starch-based hydrogel as a multifunctional SEI for ultra-stable and flexible zinc-ion batteries. J. Mater. Chem. A. 2025, 13, 25815-28.
241. Clarisza, A.; Bezabh, H. K.; Jiang, S. K.; et al. Highly concentrated salt electrolyte for a highly stable aqueous dual-ion zinc battery. ACS. Appl. Mater. Interfaces. 2022, 14, 36644-55.
242. Jiang, L.; Yao, L.; Wang, G.; Liu, C.; Chi, X.; Liu, Y. Long-duration aqueous Zn-ion batteries achieved by dual-salt highly-concentrated electrolyte with low water activity. J. Energy. Chem. 2025, 101, 778-85.
243. Gu, D.; Yuan, Y.; Liu, J.; et al. The electrochemical properties of bismuth-antimony-tin alloy anodes for magnesium ion batteries. J. Power. Sources. 2022, 548, 232076.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.