





Fenretinide in cancer therapy and chemoprevention: past, present and future
 0
0 
Abstract
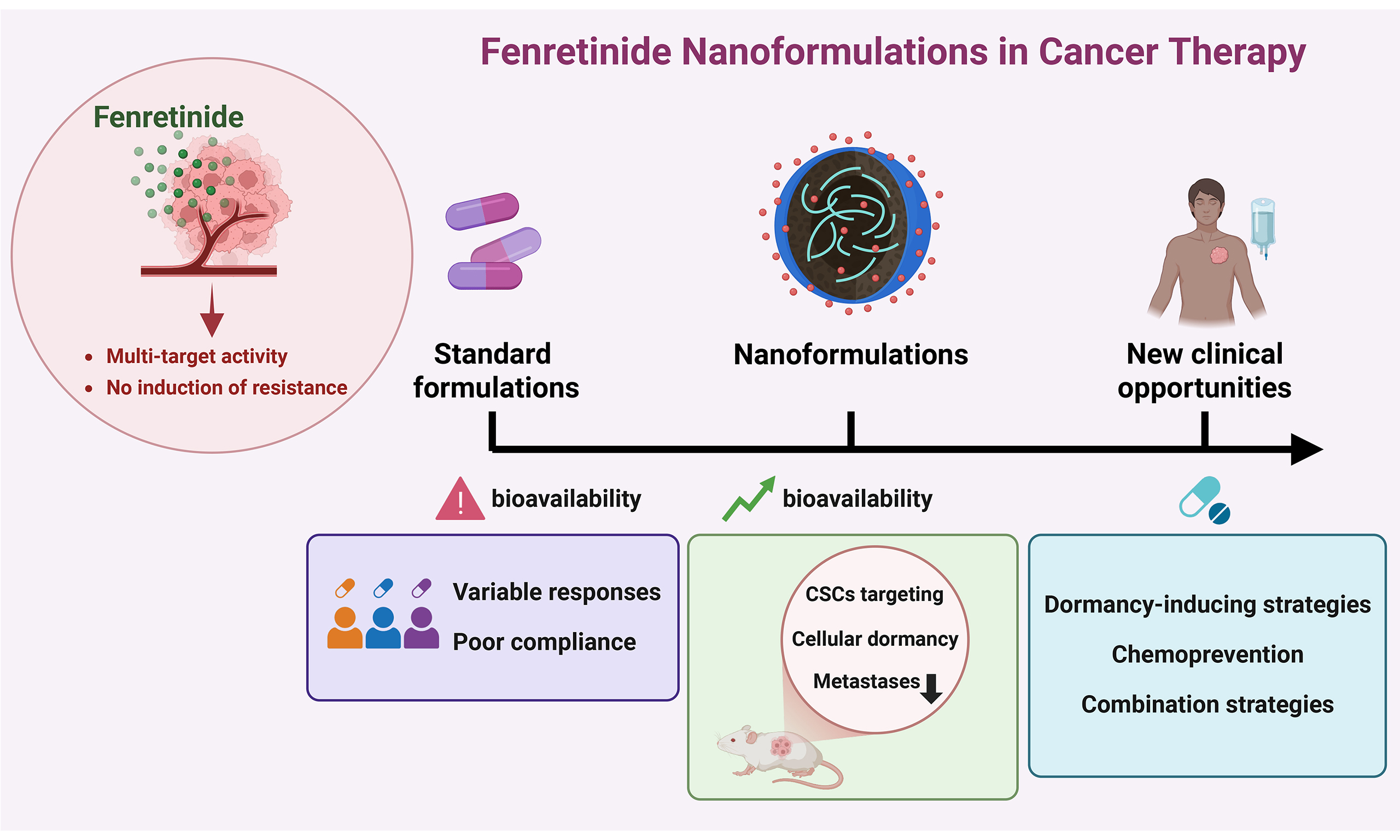
Fenretinide [N-(4-hydroxyphenyl) retinamide, 4-HPR] is a synthetic retinoid known for decades for its antitumor activity and favorable toxicological profile. In vitro and in vivo studies have demonstrated that fenretinide has broad-spectrum anticancer potential, eliciting multiple biological effects and acting as a multi-target compound that influences both tumor cells and the tumor microenvironment. However, the clinical application of fenretinide has been hampered by unfavorable physicochemical properties, such as poor solubility and low bioavailability, leading to highly variable outcomes in clinical studies. The development of novel fenretinide nanoformulations based on drug encapsulation in nanomicelles has helped overcome bioavailability limitations and improve anticancer efficacy in preclinical studies, thereby paving the way for new clinical opportunities. Moreover, recent studies have shown that fenretinide restrains the cancer stem cell compartment and promotes tumor cell dormancy, suggesting its potential use in dormancy-inducing strategies aimed at preventing and/or controlling metastatic disease. Here, we provide an overview of fenretinide’s mechanisms of action in cancer cells, summarize the available clinical evidence and discuss advances achieved with novel formulations. Finally, we discuss the potential integration of fenretinide at different stages of clinical management, with particular emphasis on its prospective use as a chemopreventive strategy or as a therapeutic option for disease control.
Keywords
INTRODUCTION
Retinoids are a class of natural and synthetic compounds structurally related to vitamin A. They have long been studied for their effects on crucial biological processes, such as cell growth, differentiation and metabolism. In addition, retinoids have a well-established reputation for their therapeutic potential across various tumor types, primarily through the modulation of nuclear retinoic acid receptors (RARs) and retinoid X receptors (RXRs), resulting in growth inhibition and induction of cancer cell apoptosis. Moreover, retinoids have attracted attention for their ability to promote cellular differentiation in multiple tumor types[1,2]. Notably, the concept of differentiation therapy originated from the observation that, in acute promyelocytic leukemia (APL), leukemic blasts can be induced to differentiate into mature granulocytes following treatment with all-trans retinoic acid (ATRA)[3-5]. However, despite the success of APL treatment protocols, the broader clinical application of retinoids remains limited by their high toxicity and poor bioavailability.
Among synthetic retinoids, fenretinide [N-(4-hydroxyphenyl) retinamide, 4-HPR] has emerged as a particularly promising antitumor agent due to its unique pharmacological profile. Originally produced in the USA in 1960, this compound is a synthetic amide derived from ATRA but behaves as an atypical retinoid with both RAR-dependent and RAR-independent activities[6]. Early studies demonstrated that, unlike ATRA, which primarily promotes cellular differentiation, fenretinide acts by inhibiting cell growth and inducing apoptosis, even in ATRA-resistant cell lines[6]. Indeed, it has been demonstrated that the pro-apoptotic activity of fenretinide involves multiple RAR-independent mechanisms, including the generation of mitochondrial reactive oxygen species (ROS) and the activation of caspases [Figure 1][7,8]. Moreover, by targeting dihydroceramide desaturase 1 (DES1), fenretinide induces the accumulation of dihydroceramide (dhCer), a bioactive lipid with pleiotropic effects on several cellular functions [Figure 1][8-10]. Beyond its pro-apoptotic activity, fenretinide exerts antitumor effects by modulating key biological processes involved in cell proliferation, biosynthesis and survival, such as suppression of the mechanistic target of rapamycin (mTOR) pathway, induction of dormancy and autophagy[8,11-13]. In addition, fenretinide has attracted considerable attention for its ability to affect cancer stem cells (CSCs) [Figure 1], which play a pivotal role in tumor progression, relapse, metastasis and resistance to radio- and chemotherapy[14-18]. Interestingly, fenretinide has been reported to exhibit anti-angiogenic properties and to modulate the tumor microenvironment (TME), leading to impaired tumor growth[19-22]. The main anticancer effects of fenretinide are summarized in Figure 1 and Table 1. Cancer constitutes one of the leading causes of morbidity and mortality worldwide[23]. Conventional therapies such as chemotherapy and radiotherapy often lead to a temporary response, ultimately promoting therapy resistance and subsequent disease progression[24-27]. One of the major causes of therapy failure is intra-tumor heterogeneity[28-30], which results from the interplay between cancer cells and the TME. Cancer cells undergo continuous genetic, epigenetic and metabolic alterations that further increase tumor heterogeneity[8,24]. Moreover, intra-tumor heterogeneity evolves over time and is exacerbated in response to therapy[30,31]. This dynamic and heterogeneous nature of cancer limits the efficacy of therapies targeting a single molecular pathway, which often fail to achieve complete tumor eradication due to the high plasticity and clonal adaptability of cancer cells. Multi-target treatment strategies proposed in recent years offer several advantages, such as the ability to overcome clonal heterogeneity, to decrease the emergence of multidrug resistance and to reduce systemic toxicity[32,33]. In this context, fenretinide has emerged as a promising therapeutic candidate due to its broad-spectrum activity and its ability to simultaneously modulate multiple pathways involved in tumor progression, along with its favorable toxicological profile. Indeed, fenretinide exhibits selective cytotoxicity toward malignant cells while sparing normal tissues, thus minimizing the toxic effects typically associated with retinoids[6,12,14,18,34-37]. Moreover, fenretinide does not accumulate in the liver, implying a reduction of hepatic toxicity, as consistently demonstrated in preclinical studies[6,12,14]. In addition, fenretinide has shown well-tolerated safety profiles in early-phase clinical trials, further supporting its potential integration into combination treatment regimens[38-40]. Despite decades of evidence supporting its antitumor efficacy and favorable toxicity profile, the clinical use of fenretinide has been limited by its challenging physicochemical properties[41,42]. Unlike other retinoids such as ATRA, which achieve adequate systemic exposure despite rapid clearance, fenretinide is characterized by long elimination half-life and tissue-specific accumulation, but exhibits markedly poor bioavailability due to its lipophilicity and low aqueous solubility[43]. These pharmacokinetic limitations required the use of high-dose regimens to achieve therapeutic plasma concentrations in earlier clinical trials, thereby compromising drug tolerability and reducing patient compliance[44]. In recent years, several laboratories have sought to improve fenretinide bioavailability by developing novel formulations with improved solubility and enhanced antitumor efficacy. This review outlines the mechanisms underlying the antitumor effects of fenretinide and discusses preclinical evidence supporting its potential use in cancer treatment and chemoprevention. Finally, we summarize the clinical findings across different cancer types, highlighting advances achieved with the use of novel fenretinide formulations developed in recent years.
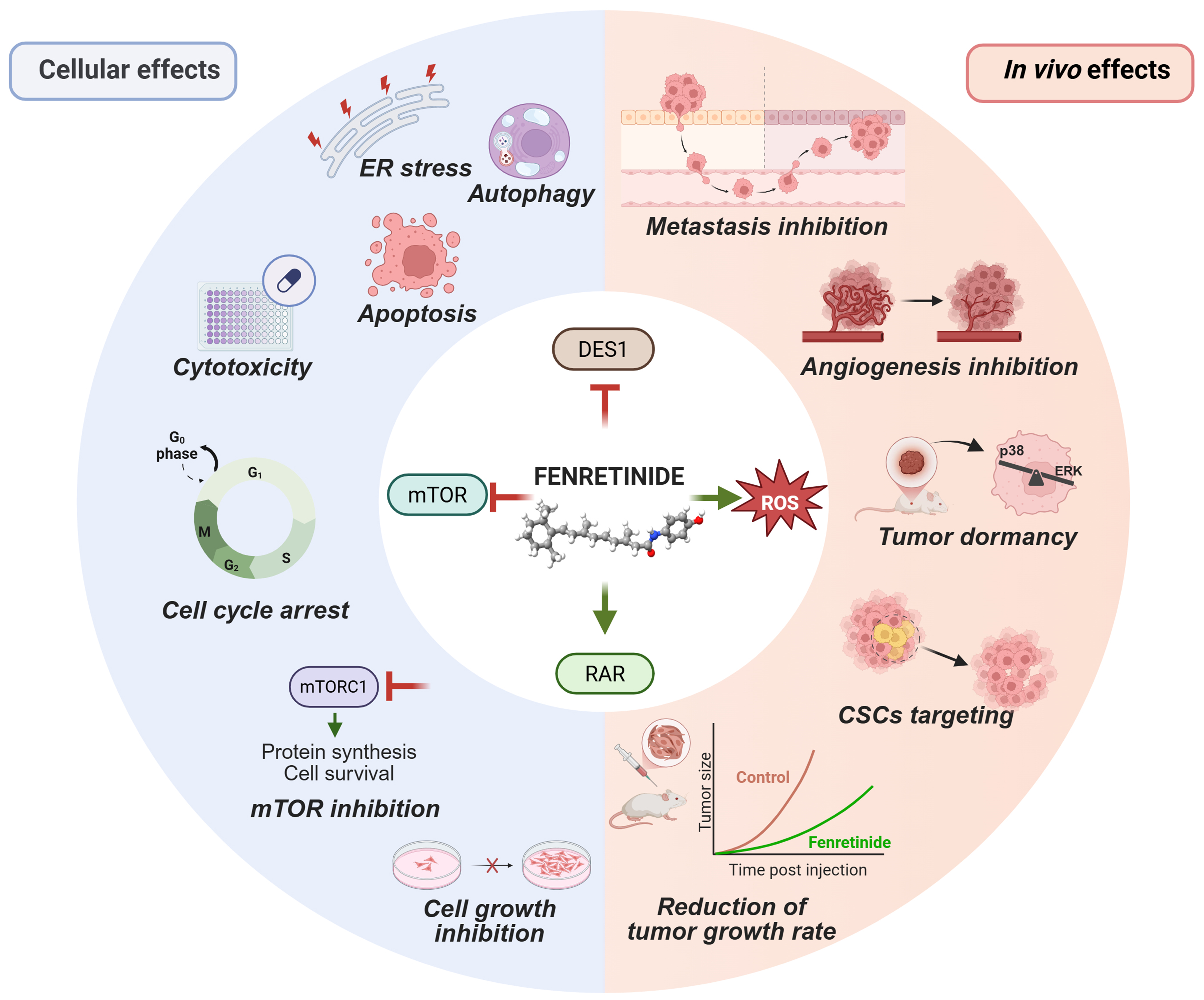
Figure 1. Multifaceted antitumor mechanisms of fenretinide. Cellular (left) and in vivo (right) antitumor effects of fenretinide are driven by multiple mechanisms (center), such as direct interaction with RAR, suppression of the mTOR pathway, generation of ROS and generation of lipid second messengers (the latter by DES1 inhibition). Red T-bars indicate inhibition. Green arrows indicate activation. Created in BioRender. Verachi, P. (2026) https://BioRender.com/5r4zhea. RAR: Retinoic acid receptor; mTOR: mechanistic target of rapamycin; ROS: reactive oxygen species; DES1: dihydroceramide desaturase 1; ER: endoplasmic reticulum; mTORC1: mTOR complex 1; ERK: extracellular signal-regulated kinase; CSCs: cancer stem cells.
Summary of antitumor effects of fenretinide
| Mechanism | Description | Key pathways/molecules |
| Apoptosis induction | Induces apoptosis through RARβ/γ | RARβ/γ, Nur77, Bcl-2 |
| Induces apoptosis through RAR-independent pathways involving ROS generation, Bcl-2 conformational change, and caspase activation | Bcl-2, ↑ROS, caspase-3 | |
| ROS generation and mitochondrial stress | Promotes ROS generation and CytC release; disrupts PKCδ-CytC signalosome | ↑ROS, CytC, PKCδ |
| Bioactive lipids modulation (Cer/dhCer) | Inhibits DES1, increases dhCer levels, triggers ER stress, autophagy, and cell cycle delay, contributing to cytotoxicity | ↓DES1, ↑dhCer, ER stress, mTORC1 |
| mTOR pathway inhibition | Directly binds to mTOR ATP pocket and inhibits mTORC1/2, reducing cell proliferation and survival signaling | ↓mTORC1, ↓mTORC2, PI3K/AKT |
| Autophagy induction | Activates autophagy in a context- and dose-dependent manner; can promote survival or cell death depending on ROS levels and apoptotic efficiency | ↑dhCer, ↑ROS, ↑UPR, ↑LC3/LC3-II, p38, PI3K/AKT/mTORC |
| Targeting CSCs | Targets CSCs by inducing ROS and ER stress, inhibiting NF-κB and Wnt/β-catenin signaling, and promoting cell cycle arrest and dormancy | ↑ROS, ↓NF-κB, ↓Wnt/β-catenin, ↑p16, ↑p-p38 |
| Cell cycle arrest/dormancy | Induces G0/G1, G1/S, or G2/M arrest depending on cell type; modulates cyclins, CDKs, p16, p-p38, and pERK to induce cellular dormancy | ↑p16, ↑p-p38, ↓pERK, ↓CDK1, ↓CDK4, ↓cyclin D1, ↓RB |
| TME modulation | Suppresses angiogenesis, inhibits M2 macrophage polarization and cell-ECM interactions | ↓VEGF/VEGFR, ↓JNK/MMP-2, ↑BMP-2/MIC-1, STAT6 |
BIOLOGICAL ACTIVITIES OF FENRETINIDE IN CANCER
RAR-dependent and independent apoptotic effects
Typical retinoids exert their antitumor activity mainly through the activation of RARs and RXRs, resulting in decreased expression of anti-apoptotic B-cell lymphoma 2 (Bcl-2) family genes[45]. In contrast, fenretinide’s pro-apoptotic effect involves diverse signaling mediators activated by RAR-independent mechanisms, such as ROS, lipid second messengers, and stress-related proteins[7-10,46]. Additional mechanisms contributing to fenretinide’s antitumor activity include induction of autophagy, cell cycle arrest and/or slowing, reduction of the CSCs content, depression of biosynthesis processes and inhibition of survival pathways[8,12-18,47]. Transactivation assays have demonstrated that fenretinide acts as a potent transactivator of RARγ and a moderate activator of RARβ, while exhibiting no interaction with RARα and RXRα. This selective activation profile may explain its specific biological activities and favorable pharmaceutical properties[48]. A study on hepatocellular carcinoma (HCC) cell lines demonstrated that the pro-apoptotic effect of fenretinide is mediated by direct activation of RARβ, whose expression also determines the susceptibility of HCC cells to fenretinide-induced apoptosis[49]. In a subsequent study, the same authors identified nuclear receptor 4A1 (Nur77) as an additional mediator of fenretinide-induced apoptosis. Specifically, in Huh7 cells fenretinide promoted the translocation of Nur77 from the nucleus to the mitochondria, allowing its binding to Bcl-2 and inducing a conformational change of this protein toward a pro-apoptotic state[50]. Accordingly, Nur77 knockdown prevented fenretinide-induced DNA double-strand breaks and caspase-3 cleavage[50]. Similarly, in acute myeloid leukemia (AML) cells, fenretinide induced the translocation of Nur77 from the nucleus to the mitochondria, where it bound Bcl-2[51]. Another study in HCC cells showed that Nur77 directly interacted with RARβ and that the induction of Nur77 expression was dependent upon the presence of
Role of lipid second messengers in fenretinide-induced cytotoxicity
Sphingolipids are membrane lipids that, besides their structural roles, function as key signaling molecules regulating cell survival, metabolism, and stress responses[59]. Ceramide (Cer), a bioactive lipid primarily produced through de novo sphingolipid biosynthesis, has attracted considerable attention for its ability to promote apoptosis in response to cellular stress[60]. In various cancer cell models, fenretinide has been shown to increase Cer levels independently of caspase activation[61,62], suggesting that it regulates de novo Cer biosynthesis and/or sphingomyelin hydrolysis. Several studies have demonstrated that fenretinide inhibits DES1, the enzyme that catalyzes the conversion of dhCers into Cer[8,9,63-66]. In human neuroblastoma SMS-KCNR cells, DES1 silencing resulted in a marked accumulation of endogenous dhCers and subsequent inhibition of cell growth with cell cycle arrest in G0/G1. Moreover, fenretinide treatment inhibited desaturase activity in a dose-dependent manner without affecting messenger RNA (mRNA) or DES1 protein levels, suggesting that fenretinide may act as a direct enzymatic inhibitor[63]. Consistent with these findings, treatment of lung and colorectal cancer (CRC) spheroid cultures with fenretinide led to significant accumulation of d18-dhCer, while pre-treatment with myriocin, an inhibitor of de novo sphingolipid biosynthesis, partially protected cells from fenretinide-induced cell death[8]. Furthermore, a lipidomic study has shown that fenretinide-induced dhCers accumulation promotes autophagy by inducing autophagosome formation in prostate cancer cells[64]. Subsequent studies demonstrated a role of dhCers in autophagy by showing that elevated dhCers levels lead to delayed G1/S cell cycle progression and endoplasmic reticulum (ER) stress induction[60]. In line with this, a study on glioma cells revealed that DES1 inhibition increases the dhCer/Cer ratio in the ER membrane, thereby triggering ER stress and inhibiting the protein kinase B (Akt)/mTOR complex 1 (mTORC1) pathway, ultimately leading to autophagy[67].
Fenretinide-mediated mTOR suppression and downstream biological effects
The serine/threonine kinase mTOR is crucial for the regulation of cell survival, metabolism, cell growth, and protein biosynthesis, and is aberrantly upregulated in more than 70% of cancers, contributing to tumorigenesis[68]. Previous studies have shown that fenretinide targets the phosphoinositide 3-kinase (PI3K)/Akt/mTOR signaling pathway and that Akt phosphorylation at Ser473 is implicated in fenretinide-induced apoptosis[69,70]. In 2012 mTOR was identified for the first time as a direct molecular target of fenretinide in both in vitro and in vivo models[11]. Specifically, molecular docking analyses showed that fenretinide binds to the ATP-binding pocket of mTOR, forming hydrogen bonds with Ser2165 and Lys2187, as well as hydrophobic interactions with key residues for the binding of ATP. Direct inhibition of mTOR suppressed the activity of both mTORC1 and mTOR complex 2 (mTORC2), resulting in reduced proliferation of lung cancer cells in vitro and decreased tumor growth in mice[11]. Interestingly, mTOR knockout in A549 cells decreased their sensitivity to fenretinide, suggesting that mTOR signaling is essential for their antitumor activity. However, no obvious apoptosis was observed in JB6 C141 cells and A549 cells after exposure to fenretinide at concentrations required to inhibit mTOR and cell proliferation, suggesting that mTOR targeting by fenretinide primarily contributes to its antiproliferative activity rather than to induction of apoptosis[11]. Consistent with those findings, a recent study on human epidermal growth factor receptor 2 (HER2)/neu (neuT) transgenic mice, a model of spontaneously metastatic breast cancer (BC), demonstrated that fenretinide treatment decreased mTOR pathway activation and cell cycle progression, inhibiting the growth of primary and metastatic tumors in the absence of overt apoptosis[12].
Autophagic signals induced by fenretinide: a double-edged sword?
Autophagy is a tightly regulated catabolic process characterized by the degradation of cellular components, reorganization of intracellular membranes and vesicles, and lysosomal activity[71]. In recent years, autophagic cell death has been increasingly recognized as a key component of cellular stress responses and tumorigenesis. Indeed, cancer cells can activate autophagy as a survival mechanism in response to stressors such as radiation and chemotherapy, thus contributing to chemoresistance[13]. However, autophagy can also promote cell death, as excessive autophagic activity can lead to the digestion of the entire cell[72]. As mentioned in section “Role of lipid second messengers in fenretinide-induced cytotoxicity”, fenretinide inhibits DES1, leading to increased dhCer levels. Mechanistically, alterations in dhCer levels modify the lipid composition of intracellular membranes, triggering stress responses, such as oxidative and ER stress, that accompany or precede autophagy signals[60]. However, whether fenretinide-induced autophagy is a pro-survival or a pro-death response remains a matter of debate. Fenretinide’s ability to induce autophagy is context-dependent, with some studies supporting pro-survival effects and others linking autophagic signals to cytotoxic effects. For example, a study showed that fenretinide-induced ROS production led to the unfolded protein response (UPR) in cancer cells, and the efficiency of this response determined cell fate. An impaired UPR leads to insufficient autophagosome formation and promotes apoptosis, whereas a complete UPR response enhances autophagic clearance of protein aggregates, favoring cell survival[73]. Fenretinide has been reported to trigger autophagic cell death when apoptotic pathways are deregulated, as in the case of caspase-3-defective Michigan Cancer Foundation-7 (MCF-7) cells. Conversely, an isogenic cancer cell line with functional caspase-3 did not exhibit autophagy features following fenretinide treatment[13]. In CRC and lung cancer spheroids, fenretinide treatment increased the expression of both microtubule-associated protein 1A/1B-light chain 3 (LC3) and its lipid-modified form LC3-II, and the autophagy inhibitor 3-methyladenine attenuated its cytotoxic effects, indicating that autophagic signals contribute to fenretinide-induced cell death in these models[8]. A recent review has proposed that the cell fate balance between apoptosis and autophagy following fenretinide exposure is dose-dependent and strictly linked to ROS levels. High concentrations of fenretinide induce elevated ROS levels, activating the p38-dependent apoptotic pathway and suppressing the PI3K/AKT/mTORC, DES1 and RARβ-Nur77 pathways, whereas lower concentrations lead to a modest increase of ROS levels, activating autophagy pathways[41].
Fenretinide and CSCs
CSCs were first identified in pioneering studies on AML, which demonstrated that only a rare subset of leukemic cells expressing stemness-associated markers was capable of initiating disease in immunodeficient mice[74,75]. Subsequent studies identified CSCs in a variety of solid tumors, including brain[76], prostate[77], colorectal[78], pancreatic[79], breast[80] and lung cancer[81]. CSCs are characterized by self-renewal capacity, tumor-initiating potential and intrinsic resistance to cytotoxic agents, and therefore constitute key drivers of tumor relapse and therapeutic failure[24]. They can evade the toxic effects of chemotherapy through a variety of mechanisms, including activation of pro-survival pathways such as PI3K/Akt/mTOR, upregulation of anti-apoptotic factors, and entry into a chemoresistant state of dormancy[24,82,83]. Moreover, it has been demonstrated that CSCs can exploit autophagy as a response to stress, thereby promoting chemoresistance and subsequent disease progression[84,85]. As mentioned in the introduction, the effectiveness of differentiating therapy was first demonstrated with ATRA in the targeted treatment of APL[4,86,87], paving the way for the development of anti-CSCs therapeutic strategies. However, it has been demonstrated that differentiated cells can revert to an undifferentiated phenotype over time, highlighting the limitations of this approach[88-91]. A recent study proposed the use of fenretinide as a potential alternative therapeutic strategy for APL in cases of resistance after protracted treatment with ATRA. In particular, the authors of this study demonstrated that the antitumor activity of fenretinide relies on apoptosis induction rather than differentiation[92]. Consistent with this mechanism, a previous study in AML showed a selective cytotoxic activity on CD34+ leukemic stem/progenitor cells (LSCs) without affecting the normal counterparts[18]. LSCs predominantly exhibit a quiescent phenotype that renders them resistant to conventional chemotherapeutic agents[18]. Notably, fenretinide was found to inhibit the clonogenic capacity of primary AML CD34+ cells in vitro and reduce the engraftment of primary and secondary recipients in vivo, suggesting a specific effect on tumor-initiating cells. Mechanistically, fenretinide induces ROS production and inhibits the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Wnt/β-catenin pathways, both of which support LSCs survival and chemoresistance[18] [Figure 2]. Similar findings have been reported in chronic myeloid leukemia, where fenretinide preferentially targets LSCs through the oxidative/ER stress-mediated pathway[15]. Additional studies have demonstrated that fenretinide is also effective against CSCs in various solid tumors, such as medulloblastoma, colon, breast, and ovarian cancer[8,16,17,47]. Using tumor spheroids enriched in stem-like cells, fenretinide was shown to preferentially target CSCs through ROS induction, ER stress, and inhibition of cell-cycle progression[16,17]. Notably, low levels of ROS in CSCs are associated with enhanced tumorigenicity and resistance to radiotherapy[93], suggesting that the stress-inducing effects of fenretinide may underlie its activity against these cells. In addition, fenretinide displayed marked efficacy against patient-derived CSCs both in vitro and in CSCs-derived xenografts of lung cancer, melanoma and CRC[8,14]. Its anti-tumor activity correlated with reduced cell proliferation, induction of apoptosis, modulation of lipid metabolism, and downregulation of CSCs-specific markers. In primary lung cancer spheroids, fenretinide decreased the expression of stemness-related genes such as NANOG, SOX2 and POU5F1, whereas in CRC spheroids it reduced Wnt signaling activity[8] [Figure 2]. In addition, reverse-phase proteomic analysis of fenretinide-treated primary lung cancer spheroids revealed a widespread repression of the mTOR pathway[8] [Figure 2]. Consistently, a recent study in neuT mice showed that treatment with fenretinide prevented both initiation and progression of metastases through a combined induction of antiproliferative signals and inhibition of the mTOR pathway[12]. Taken together, these findings strongly suggest that fenretinide preferentially targets CSCs through multiple mechanisms, including induction of ROS production, ER stress, and inhibition of key pathways that sustain CSCs survival and chemoresistance [Figure 2].
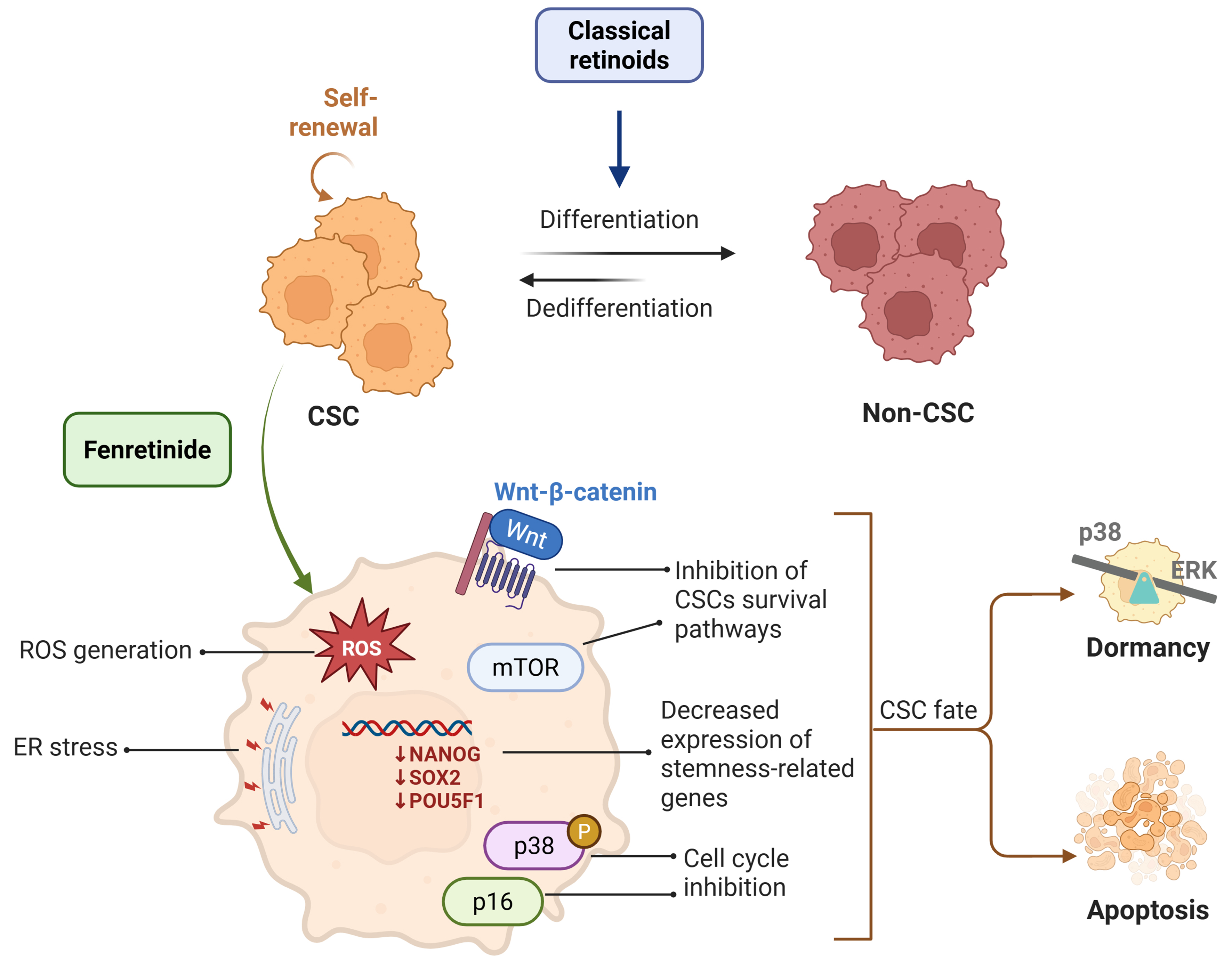
Figure 2. Mechanisms of CSC targeting by fenretinide. While classical retinoids are known to promote the differentiation of CSCs, fenretinide can reduce the content of this population by multiple mechanisms, i.e., by promoting apoptosis or inducing dormancy. Created in BioRender. Verachi, P. (2026) https://BioRender.com/qawlsjb. CSC: Cancer stem cell; ROS: reactive oxygen species; ER: endoplasmic reticulum; mTOR: mechanistic target of rapamycin; ERK: extracellular signal-regulated kinase.
Cell cycle inhibition and dormancy-related effects of fenretinide
Several studies have demonstrated that fenretinide can induce cell cycle arrest across various cancer types. However, the reported effects on the cell cycle are not consistent and appear to depend on both the experimental context and the tumor type. In a study on human CRC spheroids, treatment with fenretinide increased the proportion of cells in the G1 and G2 phases, with a corresponding decrease of the proportion of cells in the S phase. Moreover, fenretinide-treated cells exhibited downregulation of genes involved in G1/S and G2/M cell cycle transitions[16]. In medulloblastoma cells, the impact of fenretinide on the cell cycle varied depending on the dose and the cell line tested. Specifically, fenretinide induced a dose-dependent increase in the proportion of DAOY cells in the S-phase, whereas ONS-76 cells showed an increased rate of apoptosis. Consistently, treatment with fenretinide caused a downregulation of cyclin D1 and cyclin-dependent kinase 4 (CDK4) in DAOY cells, while in ONS-76 only the highest dose tested resulted in a decrease in CDK4 expression. Notably, in both cell lines fenretinide induced checkpoint kinase 2 (CHK2) phosphorylation, which was associated with cell cycle arrest in S-phase[47]. Similarly, in AML HL-60 cells fenretinide downregulated cyclin-dependent kinase 1 (CDK1) and retinoblastoma (RB) expression, while increasing phosphorylated RB (p-RB), ultimately leading to cell cycle arrest in the S-phase[51]. In A2780 ovarian carcinoma cells, treatment with fenretinide caused a modest arrest in the G1 phase without altering the expression of the G1 regulators cyclin B1, cyclin D1, and cyclin E. Interestingly, the same study demonstrated that 4-oxo-fenretinide, an active metabolite of fenretinide, was able to induce G2/M arrest and alter the expression of the cell cycle regulatory proteins CDC25C and phosphorylated CDK1 (p-CDK1)[94]. In primary lung and CRC spheroids, a novel fenretinide nanoformulation (NanoFEN) decreased the expression of cell cycle-related factors while increasing the levels of p16 and phosphorylated p38 (p-p38), suggesting a block at the G0/early G1 phase of the cell cycle [Figure 2][8]. This effect was further supported by the use of the mVenus p27K- reporter, which labels cells in the G0 phase and at the G0/G1 transition[95]. NanoFEN treatment markedly increased the number of Venus+ cells, indicating that it promotes entry into, or retention of, a quiescent state[8]. These findings were subsequently validated in vivo in neuT mice treated with an orally bioavailable fenretinide nanoformulation (Bio-nFeR)[12]. Bio-nFeR significantly reduced the growth of primary tumors and decreased the expression of the proliferation marker Ki67. Similarly, it reduced the number and size of lung metastases, which exhibited decreased levels of Ki67 and proliferating cell nuclear antigen (PCNA). In addition, primary tumors treated with Bio-nFeR showed a strongly increased expression of p-p38 and a decreased amount of phosphorylated extracellular signal-regulated kinase (pERK), where the low pERK/p-p38 ratio represents a key feature of cancer dormancy [Figure 2][96]. Treatment with Bio-nFeR also resulted in suppression of the mTOR pathway in murine breast tumors, consistent with the notion that mTOR inhibition promotes a state of cellular “hibernation” in both stem cells and cancer cells[82]. Taken together, these results indicate that in neuT mice, fenretinide counteracts tumor growth and metastatic progression by inducing cellular dormancy alongside suppression of metabolic and biosynthetic pathways[12]. In recent years, the concept of therapeutically inducing tumor dormancy has gained increasing attention as a strategy to transform cancer into a clinically manageable, non-aggressive disease[97]. Emerging preclinical and clinical evidence supports the feasibility of dormancy-based therapeutic approaches, particularly in the context of disseminated tumor cells (DTCs) and minimal residual disease[97-99]. In this framework, the ability of novel fenretinide formulations to induce tumor dormancy highlights their potential as chemopreventive agents in BC and possibly in other tumors. When considering fenretinide-induced dormancy, it is important to distinguish between cellular dormancy and tumor mass dormancy. Cellular dormancy refers to individual cancer cells entering a reversible G0/G1 arrest while maintaining metabolic activity without proliferation[1,7,8], whereas tumor mass dormancy is defined as a dynamic equilibrium between cellular proliferation and apoptosis that results in stable tumor size without net expansion[100,101]. In this context, fenretinide-induced modulation of p-p38/pERK balance, together with repression of the mTOR pathway, is consistent with the induction of cellular dormancy. Cancer cell dormancy is increasingly recognized as a clinically relevant state that enables long-term persistence of DTCs, thereby contributing to minimal residual disease and tumor relapse.
Modulation of the TME by fenretinide
A growing body of evidence shows that fenretinide is capable of modulating the TME through perturbation of signaling kinases and cell-extracellular matrix (ECM) interactions, inhibition of angiogenesis, and regulation of immune cells [Figure 3]. In a recent study on oral squamous cell carcinoma (OSCC), treatment with fenretinide induced several cancer-preventive effects including inhibition of basement membrane invasion, suppression of anchorage-independent growth, disruption of actin cytoskeletal components and inhibition of the focal adhesion kinase, a crucial protein that confers invasive properties[20]. Using molecular docking and kinase assays, it was demonstrated that fenretinide binds with high affinity to the ATP-binding pocket of all three c-Jun N-terminal kinase (JNK) isoforms, thereby inhibiting of the JNK signaling pathway associated with epithelial–myoepithelial transition. Functionally, fenretinide treatment reduced OSCC cell adhesion to the ECM and activation of matrix metalloproteinases (MMPs) [Figure 3]. In an in vivo OSCC model, local treatment with biodegradable polymeric implants containing fenretinide significantly downregulated the expression of the proto-oncogene ERG, an epithelial marker strongly associated with angiogenesis[20]. Consistent with these findings, other studies have demonstrated that fenretinide exerts robust anti-angiogenic effects through both direct and indirect mechanisms, reducing neovascularization and thereby enhancing antitumor efficacy[19]. In multiple myeloma (MM), treatment with fenretinide inhibited cell growth of cancer cells in their bone marrow microenvironment, both directly through induction of apoptosis and indirectly through inhibition of osteoclastogenesis and angiogenesis. Consistently, fenretinide inhibited tube formation by human umbilical vein endothelial cells (HUVEC) stimulated by either a cocktail of pro-angiogenic growth factors or by conditioned media derived from MM cells[102]. In a rat model of prostate cancer, fenretinide treatment significantly decreased carcinogenesis and reduced tumor vascularization[103]. Similarly, in a xenograft model using Y79 RB cells, fenretinide inhibited tumor growth through a robust reduction of microvessel formation. These findings were confirmed in in vivo Matrigel sponge assays, where fenretinide inhibited neovascularization induced by pro-angiogenic factors[104]. In Kaposi’s sarcoma, a highly vascularized tumor type, oral administration of fenretinide inhibited ectopic tumor growth and significantly reduced vascular density[105]. At the molecular level, fenretinide exerted its anti-angiogenic effects through down-regulation of the vascular endothelial growth factor (VEGF) signaling axis, a crucial pathway that drives angiogenesis. Specifically, the compound was able to reduce the expression levels of both VEGF-A and its receptor VEGFR-2 in endothelial and Kaposi’s sarcoma cells[105] [Figure 3]. Furthermore, in a mouse model of hepatocarcinogenesis VEGF-A was found downregulated in pre-neoplastic liver lesions following fenretinide treatment, thereby impairing progression of the lesions to cancer[106].
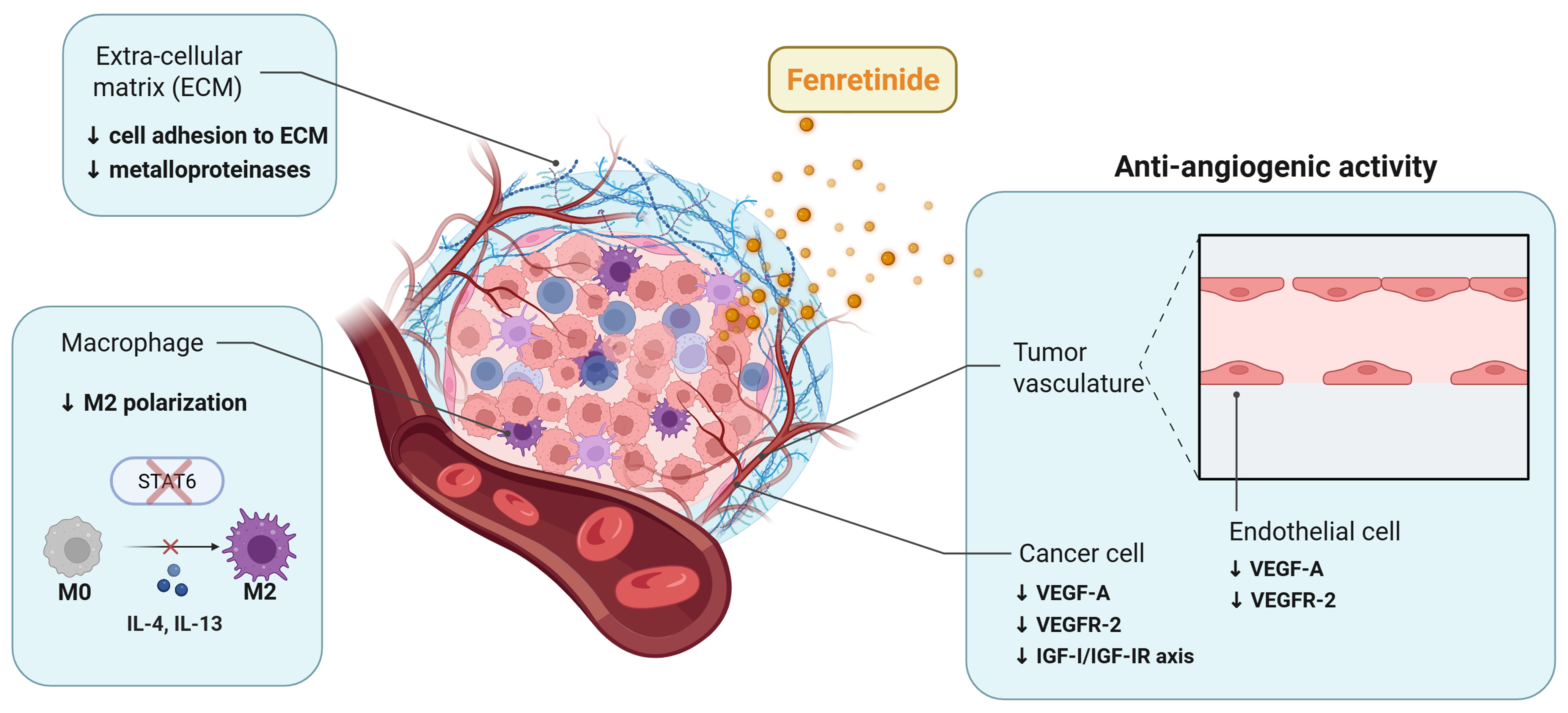
Figure 3. Modulation of the TME by fenretinide. Fenretinide shapes the TME by inhibiting angiogenesis, reducing the content of pro-tumor M2 macrophages, and disrupting tumor cell-ECM interactions. Blue boxes indicate the main effects of fenretinide on the non-cellular and cellular components of the TME. Created in BioRender. Verachi, P. (2026) https://BioRender.com/ugvfbl3. TME: Tumor microenvironment; ECM: extracellular matrix; STAT: signal transducer and activator of transcription; IL: interleukin; VEGF-A: vascular endothelial growth factor-A; VEGFR-2: VEGF receptor-2; IGF-I: insulin-like growth factor I; IGF-IR: IGF-I receptor.
Several studies have linked the anti-angiogenic properties of fenretinide to its interaction with RARs, particularly RARα and RARβ[19]. Other studies have suggested that fenretinide is able to target the insulin-like growth factor-I (IGF-I)/IGF-I receptor (IGF-IR) axis, a key pathway promoting VEGF expression via hypoxia-inducible factor (HIF)-1α signaling. Consistent with this mechanism, treatment with fenretinide decreased plasma IGF-I levels in patients with breast and bladder cancer, downregulated IGF-I and IGF-IR in BC cells in vitro and suppressed IGF-I–mediated proliferation in meningioma and RB models[19] [Figure 3]. Moreover, it has been demonstrated that fenretinide directly targets endothelial cells, interfering with multiple stages of neo-vessel formation. A mechanistic study showed that fenretinide reduces endothelial cell motility and invasiveness by inhibiting the activity of MMP-2, an enzyme essential for ECM degradation during vessel sprouting[22]. In addition, fenretinide upregulates bone morphogenetic protein-2 (BMP-2) and macrophage inhibitory cytokine-1 (MIC-1), two multifunctional cytokines with anti-angiogenic activity in vitro and in vivo. Notably, treatment with antibodies against BMP-2 counteracted the inhibitory effects of fenretinide, suggesting that suppression of angiogenesis is a key contributor to its antitumor activity[22]. Collectively, these findings indicate that fenretinide exerts a multifaceted inhibition of angiogenesis by acting on both tumor cells and components of the vascular TME while sparing the normal vasculature, as observed in clinical trials[107]. Interestingly, fenretinide has been shown to suppress IL-4/IL-13-mediated M2 polarization of RAW264.7 macrophages. This effect was associated with inhibition of phosphorylation of signal transducer and activator of transcription 6 (STAT6), a key regulator of M2 polarization [Figure 3]. Moreover, fenretinide inhibited tube formation by HUVEC cells exposed to conditioned media derived from M2 macrophages[21]. Consistent with these findings, in adenomatous polyposis coli (APC) transgenic mice, fenretinide inhibited colorectal tumorigenesis through a reduction of the M2-like macrophage population and a subsequent block of tumor angiogenesis[21]. Figure 3 and Table 1 summarize the effects of fenretinide on the TME, including its anti-angiogenic properties, inhibition of macrophage M2 polarization and suppression of cell-ECM interactions. Collectively, these data indicate that fenretinide actions create an unfavorable microenvironment for tumor progression, further supporting its potential as a chemopreventive and antitumor agent.
CLINICAL EVALUATION OF FENRETINIDE IN CANCER
The antineoplastic and chemopreventive activity of fenretinide has been validated in several animal models, supporting its potential clinical application in cancer[8,12,14,42,108]. Clinically, fenretinide has been studied in phase I-III chemoprevention and chemotherapeutic trials primarily using an oral gelatin capsule formulation containing fenretinide suspended in corn oil and polysorbate 80 (National Cancer Institute fenretinide, NCI-FeR) [Table 2]. However, the results of these studies have shown high interpatient variability, primarily attributable to therapeutically effective drug concentrations not being consistently reached within tumor tissues. Pharmacokinetic studies reported that, even with dose escalation, plasma concentrations and tumor response rates did not increase proportionally, as would have been expected[39,40,109]. The variable and insufficient absorption of fenretinide can be attributed to its unfavorable pharmacokinetic profile: the low aqueous solubility and high lipophilicity of the molecule hinder its absorption and systemic distribution, ultimately restricting its penetration into tumor tissues[44]. Cooper et al. estimated a mean oral bioavailability of 16% in beagle dogs given a single 10 mg/kg oral dose of capsules, equivalent to a human dose of approximately 200 mg/m2[42,110]. In recent years, new fenretinide formulations have been developed in order to increase plasma concentrations of the drug, thereby potentially improving clinical responses (Table 3 and section “Novel fenretinide formulations: preclinical studies and clinical evidence”).
Summary of clinical studies evaluating the effects of standard fenretinide formulations in cancer
| Formulation | Delivery method | Key advantages | Main limitations | Tumor | Daily dose | Plasma and/or intratumor concentrations | Clinical trial phase | Clinical response | Ref. |
| Soft gelatin capsule (NCI fenretinide) | Oral corn-oil and polysorbate 80 capsule | Low toxicity | Poor and highly variable bioavailability; limited efficacy; poor patient compliance | Neuroblastoma (n = 53) | 100-4,000 mg/m2 | Plasma: 1.3-12.9 µM | I | No objective response; 77% SD | [109] |
| Neuroblastoma (n = 39), Ewing sarcoma (n = 5) and other (n = 10) | 350-3,300 mg/m2 (MTD = 2,475 mg/m2) | Plasma: 9.9 µM at the MTD | I | 1 CR, 43% SD (neuroblastoma); 1 SD (Ewing sarcoma); 1 SD (melanoma) | [39] | ||||
| Advanced renal cell carcinoma (n = 19) | 1,800 mg/m2 | Intratumor: 3.6-7.9 µM | II | No objective response; 37% with SD | [111] | ||||
| Small-cell lung cancer (n = 19) | 1,800 mg/m2 | Plasma: 7.4 +/- 4.25 µM | II | No objective response; 24% with SD | [112] | ||||
| Prostate cancer (n = 23) | 1,800 mg/m2 | NA | II | No objective response; 30% with SD | [113] | ||||
| Prostate cancer (n = 27) | 1,800 mg/m2 | NA | II | No objective response | [114] | ||||
| Ovarian cancer (n = 22) | 400-800 mg | Plasma 1.4 µM at the highest dose | I-II | No objective response | [116] | ||||
| Ovarian cancer (n = 31) | 1,800 mg/m2 | Plasma: 3.1-12.5 μM | II | 42% SD at ≥ 9 μM; OS at 18 months 66% at ≥ 9 μM | [117] | ||||
| Brain tumor (n = 22 AG; n = 23 GBM) | 1,200 or 1,800 mg/m2 | Plasma: 1,213 +/- 261 ng/mL at the 900 mg/m2 bid dosage | II | 1 partial radiologic response; 29% SD (AG arm), 9% SD (GBM arm) at 6 months | [115] | ||||
| BC and melanoma (n = 31) | 300-400 mg | NA | II | Limited activity: 6.7% mixed response; 26.7% SD | [118] | ||||
| BC (n = 872 fenretinide arm vs. N = 867 observation arm) | 200 mg (5 years) | NA | III | 50% reduction in second BC risk in premenopausal women (≤ 40 years) | [38] |
Summary of preclinical and clinical studies evaluating the effects of novel fenretinide formulations in cancer
| Formulation | Delivery method | Key advantages | Main limitations | Tumor | Daily dose | Plasma and/or intratumor concentrations | Clinical trial phase | Clinical response | Ref. |
| Fenretinide/LSX | LYM-X-SORB™ lipid matrix | Increased plasma levels vs. oral capsules | Limited long-term compliance due to GI side-effects; poor patient compliance | Adults with solid tumors and lymphomas (n = 20) | 800-1,300 mg/m2 | Plasma: ≥ 10 µM on day 7 at 1,000 mg/m2/day; > 20 μM in 1 patient who received 24 cycles | I | 1 SD (cutaneous T-cell lymphoma) | [120] |
| Neuroblastoma (n = 29) | 352-2,210 mg/m2 | Plasma: 21 µM on day 6 at a dosing regimen of 1,700 mg/m2/day | I | 29% SD, 13% CR | [121] | ||||
| Fenretinide/LSX + ketoconazole | Increased plasma levels vs. Fenretinide/LSX alone | Neuroblastoma (n = 34) | Cohort 1: 1,500 mg/m2 Cohort 2: Fenretinide/LSX + 6 mg/kg ketoconazole | Plasma: 18.4 µM Cohort 2 vs. 11.7 µM Cohort 1 | I | 2 CR, 1 PR, 1 mixed response (MIBG CR), 14 SD, and 12 progressive disease | [122] | ||
| Intravenous fenretinide | Lipid emulsion (mixture of egg phospholipids, glycerin, alcohol, and soybean oil) | Circumvents GI absorption issues; enables controlled dosing | Reversible hypertriglyceridemia | Malignant solid tumors (n = 18) | 905-1,414 mg/m2 | Plasma: 7-15 µg/mL at 1,131 mg/m2 | No objective response, 28% SD | [124] | |
| Peripheral T-cell lymphoma (n = 11) | 905-1,810 mg/m2 | Plasma: 50 μM at the MTD 1,280 mg/m2/day | I | Response rate of 82%: 2 CR, 2 unconfirmed PR, 5 SD at doses ≥ 905 mg/m2/day | [125] | ||||
| ST-001 nanofenretinide | Intravenous infusion | Phospholipid suspension | Relapsed/refractory T-cell non-Hodgkin lymphoma | (n = 46) *estimated | 1.25-640 mg/m2 | NA | Ia/Ib (recruiting) | Ongoing study; clinical data not available yet | ClinicalTrials.gov ID: NCT04234048 |
| Relapsed/refractory small cell lung cancer | (n = 44) *estimated | NA | Ia/Ib (recruiting) | Ongoing study; clinical data not available yet | ClinicalTrials.gov ID: NCT06922539 | ||||
| NanoFEN | Nanomicelles (complexation with 2-hydroxypropyl β-cyclodextrin) | Increased plasma levels vs. oral capsules | Preclinical only; iv administration | Preclinical (lung and CRC xenografts) | 10 mg/kg | Plasma Cmax: 730.06 (oral) and 6,932.6 ng/mL (iv) at 5 mg/kg (single administration) | NA | NA (preclinical studies only) | [8] |
| NanoFEN + lenalidomide | Nanomicelles (phospholipids and triglyceride mixed with hydroxypropyl β-cyclodextrin) | Increased drug delivery; superior antitumor response compared to the single agent | Preclinical only; iv administration | Preclinical (human neuroblastoma xenografts) | 30 mg/kg | Plasma: 10.06 µM; intratumor: 54.47 µM | NA | NA (preclinical studies only) | [128,129] |
| Bio-nFeR | Ion-pair stabilized lipid matrix | Oral administration; increased plasma levels vs. oral capsules | Preclinical only | Preclinical (lung cancer, melanoma and colon cancer CSCs-derived xenografts; neuT mice) | 100 mg/kg | Plasma: 9.2 µM vs. 1.0 µM for capsules; intratumor: 9.6 µM vs. 1.5 µM for capsules | NA | NA (preclinical studies only) | [12,14] |
Clinical studies with oral fenretinide capsules
Neuroblastoma
In a phase I trial, oral fenretinide capsules were administered to children with neuroblastoma once daily in a range of doses from 100 to 4,000 mg/m2 for 28 days followed by a seven-day drug holiday. Although no severe toxicity was observed at the highest dose, excessive amounts of corn oil caused intestinal discomfort and precluded additional dose escalation. Day 28 plasma levels ranged from 1.3 to 12.9 µM in a dose-dependent manner. While no partial or complete responses (CR) were observed, 77% of patients achieved stable disease (SD) with a median of 23 months, and some of them showed partial regression[109]. The Children’s Oncology Group reported the results from a phase I clinical trial of oral fenretinide in 54 children with high-risk solid tumors, including neuroblastoma (n = 39), Ewing sarcoma (n = 5)[39]. Doses ranging from 350 to 3,300 mg/m2 were tested for days 1 to 7 every three weeks. The maximum tolerated dose (MTD) of 2,475 mg/m2 yielded a plasma concentration of 9.9 µM. One CR and 13 patients with SD were observed among 30 assessable neuroblastoma patients. Similar outcomes were observed in one of five Ewing sarcoma patients and one melanoma patient[39].
Renal cell carcinoma
In a phase II trial, patients with unresectable or metastatic renal cell carcinoma received 900 mg/m2 twice daily (1,800 mg/m2 per day) for seven days in a three-week cycle for a total of 76 cycles. While no objective responses were observed, 37% of patients achieved SD with a median duration of 5.8 months. Consistent with the limited activity observed in patients, intratumor levels of fenretinide measured in three patients (3.6-7.9 µM) were at the lower end of the therapeutic window[111].
Small-cell lung cancer
In 19 patients with small-cell lung cancer receiving a dose of 900 mg/m2 twice daily, no objective responses were observed. However, four of 17 response-evaluable patients (24%) achieved SD after 2-17 cycles. Among 14 patients, the mean plasma concentration of fenretinide on day seven of cycle one was 7.4 µM[112].
Prostate cancer
In a phase II trial of oral fenretinide in patients with biochemically recurrent prostate cancer, the compound was administered at a dose of 900 mg/m2 twice daily for one week every three weeks for one year. The primary endpoint was a prostate-specific antigen (PSA) response, defined as a reduction of ≥ 50% and
Brain cancer
In a phase II study conducted in adults with recurrent malignant gliomas [22 patients with anaplastic gliomas (AG) and 23 patients with glioblastoma (GBM)], oral fenretinide was administered on days one to seven and 22 to 28 of six-week cycles at doses of 600 or 900 mg/m2 twice daily (1,200 and 1,800 mg/m2 per day, respectively). The trial was discontinued after the first stage due to the limited activity at the tested doses. However, 29% of patients in the AG arm and 9% of patients in the GBM arm achieved SD at six months, and one patient with AG receiving 1,800 mg/m2 achieved a partial radiologic response[115].
Ovarian cancer
An early phase I-II trial of oral fenretinide capsules (400-800 mg/day) in patients with ascitic ovarian cancer administered for up to four weeks prior to surgery found no objective responses. Mean plasma concentrations reached a mean of 1.4 µM at the highest dose tested, while drug levels in malignant ascitic cells and tumor tissue were 50 and five times lower, respectively, than those observed in carcinoma cells exposed to 1.4 µM fenretinide[116]. In a subsequent phase II trial for recurrent ovarian cancer, 1,800 mg/m2 of fenretinide (divided into twice-daily dosing) did not produce objective responses; however, 42% of patients achieved SD for a median of 7.9 months. Plasma fenretinide concentrations ranged from 3.1 to 12.5 μM (n = 24). Clinical outcomes were positively associated with fenretinide plasma levels: progression-free survival (PFS) at six months was 42% in patients achieving plasma concentrations ≥ 9 μM, compared to 17% in those below this threshold, while overall survival (OS) at 18 months was 66% vs. 13%, respectively[117].
BC
An early phase II study on 31 patients with advanced BC or melanoma treated with daily fenretinide capsules (300-400 mg/day) reported no partial or CRs[118]. In 1987, a large multicenter phase III trial conducted a long-term analysis of the efficacy of a 5-year treatment with fenretinide (200 mg per day) in reducing contralateral or second ipsilateral BC in 2,867 patients aged 30-70 years with early BC who received no systemic therapy after primary treatment. After 8 years, no difference was observed in contralateral or ipsilateral BC; however, a post-hoc analysis suggested a significant correlation between treatment and patient menopausal status (or age), showing a 35% reduction in second BC risk among premenopausal women (or those aged < 50 years) and an opposite trend in postmenopausal women (or those aged > 50 years)[38]. A median 14.6 years follow-up from 1,739 women aged 30-70 (872 in the fenretinide arm and 867 in the observation arm) demonstrated a marked chemopreventive effect in the premenopausal subgroup: among women aged ≤ 40 years the risk in second BC decreased by around 50% and the protective benefit persisted for more than a decade after treatment cessation[38]. In summary, although oral fenretinide capsules exhibited low toxicological profile and favorable tolerability, the poor solubility of the compound severely limited its systemic distribution. In most clinical studies plasma and/or intratumor concentrations of fenretinide remained below 10 µM, whereas preclinical data suggest that steady-state concentrations exceeding 10 µM are required to ensure biological activity and therapeutic efficacy[42]. However, dose escalation in pharmacokinetic studies was constrained by capsule burden and by poor patient compliance, prompting the development of improved formulations.
Novel fenretinide formulations: preclinical studies and clinical evidence
Oral fenretinide/LSX
To improve the oral delivery of fenretinide, the fenretinide was incorporated into a LYM-X-SORB lipid matrix, a mixture of lysophosphatidylcholine, monoglyceride and free fatty acids specifically designed to improve the absorption of lipophilic drugs through the proximal intestine into the lymphatic system[119]. In vivo studies have demonstrated that fenretinide/LYM-X-SORB (Fen/LXS) oral powder reached significantly higher concentrations in both plasma and tumor tissue of BALB/c mice as compared with NCI-FeR[119]. Furthermore, Fen/LXS oral powder extended survival in two of three mice bearing human neuroblastoma xenografts, further supporting its clinical utility[119]. In a phase I dose-escalation study, 20 adults with refractory malignancies received Fen/LXS oral powder administered in two divided doses for seven consecutive days every 21 days. The MTD regimens were 1,000 mg/m2/day (when given as two daily doses) and 800 mg/m2/day (when given as three daily doses) for seven days every 21 days. However, seven of 20 patients discontinued treatment due to gastrointestinal side effects or poor palatability. Plasma drug levels exhibited interpatient variability across all the doses tested, with some patients reaching concentrations higher than 9 μM[120]. In a separate phase I study, Fen/LXS was administered to 32 patients with relapsed/refractory neuroblastoma at doses ranging from 352 to 2,210 mg/m2/day. At a dose of
Intravenous lipid emulsion of fenretinide
To overcome the poor patient compliance associated with oral fenretinide, an intravenous (iv) formulation was developed using a lipid emulsion composed of a mixture of egg phospholipids, glycerin, alcohol, and soybean oil. This formulation achieved significantly higher mean plasma concentrations of fenretinide in animal studies[110]. In a phase I study involving patients with malignant solid tumors, the iv formulation was administered as a continuous infusion for five consecutive days in 21-day cycles[124]. The formulation exhibited a manageable safety profile and reached significantly higher plasma steady-state concentrations of the active metabolite compared to capsule formulations. Although no objective responses were observed, five patients achieved SD lasting for 11 to 22 weeks[124]. In hematological malignancies, iv fenretinide
Fenretinide nanoformulations: the road to the future?
Recently, novel nanoformulations based on phospholipid–liquid triglyceride mixtures have been developed to improve fenretinide delivery and bioavailability. Nanoformulations offer several advantages over traditional drug delivery methods. In tumors characterized by abnormal vasculature, intravenously administered nanoparticles circulate systemically and retain the drug payload until extravasation occurs through discontinuities of the endothelium of tumor capillaries, a process known as enhanced permeability and retention effect, leading to selective drug accumulation and release within tumor tissue. When administered orally, nanoparticles enhance solubilization and improve absorption of lipophilic drugs in the gastrointestinal tract, thus increasing systemic bioavailability and therapeutic efficacy[8]. In 2019, Orienti et al. developed a new fenretinide nanoformulation (NanoFEN) through salification and complexation with 2-hydroxypropyl β-cyclodextrin, a solubilizing excipient characterized by favorable biodistribution and minimal toxicity[8]. In mice, a single administration of NanoFEN (5 mg/kg) by oral or iv routes resulted in significantly higher plasma concentrations of fenretinide (Cmax: 730.06 and 6,932.6 ng/mL, respectively) as compared to those achieved with an equivalent dose of oral fenretinide capsules (Cmax: 298.13 ng/mL)[8]. In the same study, NanoFEN exerted a potent antitumor activity in cell lines derived from solid and hematologic tumors, in primary spheroid cultures and in xenograft models. Importantly, treatment with NanoFEN significantly inhibited the growth of lung and CRC xenografts, in the latter independently of the mutational status of the tumor cells. Moreover, a prolonged 70-day treatment in mice bearing lung cancer xenografts resulted in complete arrest of tumor growth, with no detectable signs of toxicity[8], supporting the potential use of fenretinide as a dormancy-inducing strategy. Subsequently, the same group generated Bio-nFeR, a nanomicellar fenretinide formulation with the key advantage of enabling oral administration as a liquid suspension[14]. An extensive pharmacokinetic study revealed plasma concentrations of fenretinide 1.3 and 1.6 times higher with Bio-nFeR than with NCI-FeR oral capsules after initial and repeated dosing, respectively[14]. Moreover, chronic administration of Bio-nFeR resulted in a 40% increase in plasma concentrations as compared to day one[14]. In the same study, the antitumor activity of Bio-nFeR was assessed in patient-derived three-dimensional cultures and in xenograft models, in comparison with NCI-FeR. Bio-nFeR induced cytotoxicity in cancer cells even at low concentrations, while NCI-FeR produced only a moderate reduction in cell viability at the highest dosage tested. Consistently, Bio-nFeR significantly suppressed the growth of lung cancer xenografts, whereas NCI-FeR did not affect tumor growth. Finally, no signs of systemic toxicity were observed following repeated Bio-nFeR administration, supporting the feasibility of dose escalation in further preclinical studies[14]. In xenograft models derived from lung, CRC and melanoma CSCs, Bio-nFeR caused a significant arrest of tumor growth without evidence of systemic toxicity. In conclusion, Bio-nFeR achieved plasma concentrations above the therapeutic threshold and higher than those reported in other studies. This enhanced bioavailability was accompanied by superior antitumor efficacy in vivo, providing a strong rationale for the clinical translation of Bio-nFeR[14]. In a recent study using neuT mice, treatment with Bio-nFeR significantly delayed tumor onset and, most importantly, showed high efficacy against metastatic progression, inhibiting both metastasis initiation and expansion. These findings support the potential of Bio-nFeR for both prevention and treatment of metastatic BC[12]. Notably, the doses of Bio-nFeR used in animal models were comparable to the lower doses used in drug escalation clinical trials using other fenretinide formulations[12,14]. Specifically, 100 mg/kg in mice corresponds to 300 mg/m2 in humans, whereas clinical trials using other fenretinide formulations have safely tested doses up to
Potential mechanisms of fenretinide resistance
There is no clear evidence to date that cancer cells develop resistance to fenretinide. While resistance to ATRA is well documented and involves diverse mechanisms, such as RARα mutations and activation of compensatory survival pathways, fenretinide has shown efficacy even in ATRA-resistant tumor cells[130]. Nevertheless, potential adaptive or intrinsic mechanisms of resistance to fenretinide should be considered and evaluated in future studies. Such mechanisms may involve:
(a) Alterations in the apoptotic protein network, such as upregulation of anti-apoptotic Bcl-2 family proteins or suppression of Bcl-2 homology 3 domain (BH3)-only proteins. In this regard, preclinical studies on the synergistic activity of fenretinide in combination with BH3 mimetics suggest diverse and complementary actions of the two drug types[131,132].
(b) Enhanced expression of ROS scavengers. Interestingly, a study on tumors of the Ewing sarcoma family demonstrated that the intracellular glutathione antioxidant system is a critical determinant of cellular sensitivity to fenretinide[133].
(c) Bypassing mTOR inhibition. Since fenretinide inhibits mTOR activity, mechanisms of resistance to mTOR inhibitors, including activation of compensatory kinases, feedback activation of PI3K/AKT signaling or mTOR mutations affecting drug binding[134], may theoretically block fenretinide effects on the mTOR pathway.
(d) Drug efflux mechanisms. Multidrug resistance efflux transporters and other mechanisms such as membrane rigidification can regulate intracellular fenretinide levels[92]. However, in Ewing sarcoma cells fenretinide is not a substrate of the ATP-binding cassette (ABC) transporter multidrug resistance-associated protein 1 (MRP1), underscoring tumor–specific differences in the contribution of drug efflux proteins to fenretinide sensitivity[135].
In summary, while the multifaceted mechanism of action of fenretinide makes the development of resistance less likely, tumor cells may theoretically still acquire escape mechanisms. Future studies evaluating novel fenretinide formulations will be crucial to identify and overcome potential resistance mechanisms.
CONCLUSIONS AND FUTURE PERSPECTIVES
Fenretinide has long been investigated for its pleiotropic antitumor properties and favorable toxicity profile. Unlike classic retinoids, which primarily act through nuclear RARs, fenretinide exerts its activity through both RAR-dependent and RAR-independent mechanisms, resulting in a broad spectrum of biological effects, including induction of apoptosis, ROS production and inhibition of biosynthetic pathways. Notably, fenretinide has been consistently shown to display marked anti-proliferative activity and the ability to reduce the population of stem-like cells, which are increasingly recognized as major drivers of therapeutic resistance and disease recurrence. More recently, preclinical studies in animal models of metastatic BC have shown that fenretinide is able to induce cellular dormancy, thereby suppressing both the initiation and progression of metastases. Despite extensive preclinical evidence supporting its efficacy, early clinical trials with oral formulations demonstrated only limited antitumor activity in multiple solid and hematological tumors. This discrepancy between preclinical observations and clinical data is likely attributable to pharmacokinetic limitations, as multiple studies have demonstrated that plasma and/or intratumoral concentrations of fenretinide achieved with the standard formulation often failed to reach the threshold required for cytotoxic activity. As previously mentioned, fenretinide exhibits physicochemical properties distinct from those of other retinoids such as ATRA, including marked lipophilicity, which limits its bioavailability. This difference in pharmacokinetic behavior highlights the particularly critical challenge posed by fenretinide’s limited systemic exposure and underscores the need for advanced formulation strategies to improve its clinical efficacy. In recent years, novel formulations based on lipid-based oral suspensions and nanoencapsulated delivery systems have significantly improved fenretinide delivery and bioavailability, thus renewing interest in its clinical applications. By reducing dose requirements, nanoformulations may also mitigate gastrointestinal adverse effects and improve patient compliance, which were limiting factors in early clinical trials. Consistent with this, preclinical in vivo studies using fenretinide nanoformulations have reported absence of weight loss, inappetence or other signs of gastrointestinal distress, suggesting improved tolerability as compared with earlier oral formulations[12,14]. Moreover, early-phase clinical evaluation of ST-001 nanofenretinide has demonstrated an excellent tolerability profile with no unexpected toxicities reported to date. However, systematic assessment of gastrointestinal safety in patients is still lacking[136]. Future strategies, such as the addition of flavoring agents, could further enhance the palatability of oral fenretinide formulations and facilitate their clinical translation. The recent development of novel fenretinide formulations offers a promising opportunity to facilitate its clinical translation. The multifaceted pharmacological activity of fenretinide supports its potential integration at multiple stages of cancer management. Figure 4 illustrates possible applications of fenretinide nanoformulations to cancer treatment schedules, specifically (i) chemoprevention in tumors with high risk of relapse after surgery and/or chemotherapy/radiotherapy; (ii) disease stabilization in progressing tumors; and (iii) intercalation schedule between chemotherapy cycles. First, the use of fenretinide as a chemopreventive agent is consistently supported by data from various preclinical models demonstrating its ability to promote cell cycle arrest and maintain tumor cells in a non-proliferative cell cycle state. Dormant micro-metastases or single dormant DTCs are widely recognized as key contributors to late relapse in tumors with high risk of recurrence, such as BC[137]. Although the majority of BC patients are diagnosed at an early, potentially curable stage, 20%-30% eventually experience recurrence due to occult minimal residual disease[137]. Subsets of patients at high risk of late relapse can be identified through specific patterns of genomic copy-number alterations and gene expression[138], making them eligible for therapies aimed at preventing disease recurrence. The importance of targeting dormant cancer cells to prevent BC recurrence is illustrated by a recent randomized phase II trial using hydroxychloroquine, the mTOR signaling inhibitor everolimus or their combination in BC survivors within 5 years of diagnosis who had detectable DTCs in bone marrow aspirate[139]. The ability of fenretinide to induce a dormancy phenotype highlights its potential as a chemopreventive strategy in cancer patients following surgical tumor resection with the aim of reducing the risk of disease recurrence. This concept is reinforced by a recent study showing that the novel formulation Bio-nFeR inhibited metastatic progression in neuT mice[12]. In addition, clinical trials using conventional oral formulations have shown a significant reduction in the risk of second BC in premenopausal women, with the protective effect persisting after treatment discontinuation[38]. These observations highlight a potential window of opportunity to evaluate modern nanoformulations in the adjuvant or post-adjuvant setting, where the primary objective is long-term suppression of minimal residual disease. This approach reflects the evolving paradigm of reducing recurrence risk by targeting specific biological features of dormant cancer cells. Moreover, the low-toxicity profile of fenretinide supports its suitability for chronic administration, a critical consideration in malignancies such as BC, where late relapse may occur 20-30 years after primary diagnosis[140]. Second, fenretinide may be considered for the management of advanced and rapidly progressing tumors with limited or no therapeutic options, where disease stabilization represents a clinically relevant endpoint. The historical observation that treatment with fenretinide often resulted in SD rather than in objective responses across multiple tumor types may actually represent a therapeutic goal in this context. Furthermore, the favorable safety profile of fenretinide makes this compound suitable for cancer patients debilitated by both tumor burden and prior intensive pharmacological or radiation therapies, rendering them more vulnerable to treatment-related toxicities. Third, we propose that novel fenretinide nanoformulations could be integrated into chemotherapy regimens, either in combination or in alternation to conventional cytotoxic agents. Substantial evidence indicates that standard chemotherapy, while restraining the expansion of responsive tumors, paradoxically enriches for CSCs that are responsible for chemoresistance and relapse[24]. In this context, the ability of fenretinide to target CSCs and downregulate stemness-related genes may be exploited in a clinical setting to prevent chemotherapy-induced progression. Based on this hypothesis, integration of fenretinide between chemotherapy cycles would ideally function as a “biological brake”, counteracting the rebound effects of chemotherapy and extending the durability of clinical benefit. Although not yet validated in the clinic, this hypothesis is supported by substantial preclinical evidence and warrants systematic evaluation in future studies. Indeed, several preclinical studies have already demonstrated that fenretinide can enhance the efficacy of conventional chemotherapeutic and immunotherapeutic agents. For example, Formelli et al. showed that fenretinide enhances cisplatin sensitivity in both cisplatin-sensitive and cisplatin-resistant ovarian tumor models[141]. Similarly, Kalemkerian et al. reported that fenretinide in combination with cisplatin, etoposide, or paclitaxel produced more-than-additive growth inhibition of a small cell lung cancer cell line, indicating a potential pharmacological synergy[142]. Other preclinical studies have shown that fenretinide in combination with bortezomib, selenite, ABT-263 (Navitoclax), histone deacetylase inhibitors or CD20-directed antibodies exhibited enhanced efficacy in multiple cancer types including melanoma, ovarian cancer, neuroblastoma and hematologic malignancies. Compared to single agents, drug combinations resulted in increased apoptosis, ROS accumulation, mitochondrial dysfunction and suppression of survival pathways[143-150]. Recently, Orienti et al. have shown complete regression of tumor growth in neuroblastoma xenograft models treated with fenretinide nanomicellar formulations combined with the immunomodulatory drug lenalidomide[128,129]. These findings suggest that fenretinide may synergize with immunomodulatory agents by remodeling the TME, thereby potentiating anti-angiogenic effects and increasing tumor cell susceptibility to immune-mediated responses. In this context, investigating a potential synergy with immune checkpoint inhibitors may be of interest to further increase tumor cell susceptibility to immune-mediated cytotoxicity. Despite promising preclinical evidence, clinical evaluations of traditional fenretinide formulations[151-153] in combination therapies have shown acceptable tolerability but limited efficacy, reflecting suboptimal systemic exposure. The development of novel fenretinide formulations may offer a promising strategy for overcoming these limitations by achieving higher and more sustained drug levels, thereby potentially amplifying synergistic interactions with other chemotherapeutic agents. Translating the potential of combination therapies into clinical practice will require additional preclinical studies, careful design of intercalated schedules and dosing optimization for fenretinide nanoformulations. Furthermore, the identification of biomarkers potentially predictive of response to fenretinide, such as mTOR pathway hyperactivation, expression of CSCs markers or expression of factors specifically associated to lipid metabolism, could enable stratification of patients eligible for fenretinide treatment, thus maximizing clinical benefits. In conclusion, this review highlights the pleiotropic biological effects of fenretinide and underscores the potential clinical relevance of its novel formulations. The preclinical and clinical evidence discussed herein supports the view that fenretinide should not be considered a conventional cytotoxic drug but rather a disease-modifying agent capable of attenuating tumor aggressiveness by promoting dormancy and reducing stemness. This perspective aligns with the evolving paradigm in modern oncology that emphasizes chronic disease management and therapeutic strategies targeting minimal residual disease. Taken together, fenretinide’s unique pharmacological profile together with the development of novel formulations that improve bioavailability, encourage a clinical reassessment of this drug in both therapeutic and chemopreventive settings. Novel fenretinide formulations may potentially address unmet clinical needs such as relapse prevention in high-risk patients or the management of advanced or metastatic tumors, filling a gap where conventional therapeutic approaches provide limited results.
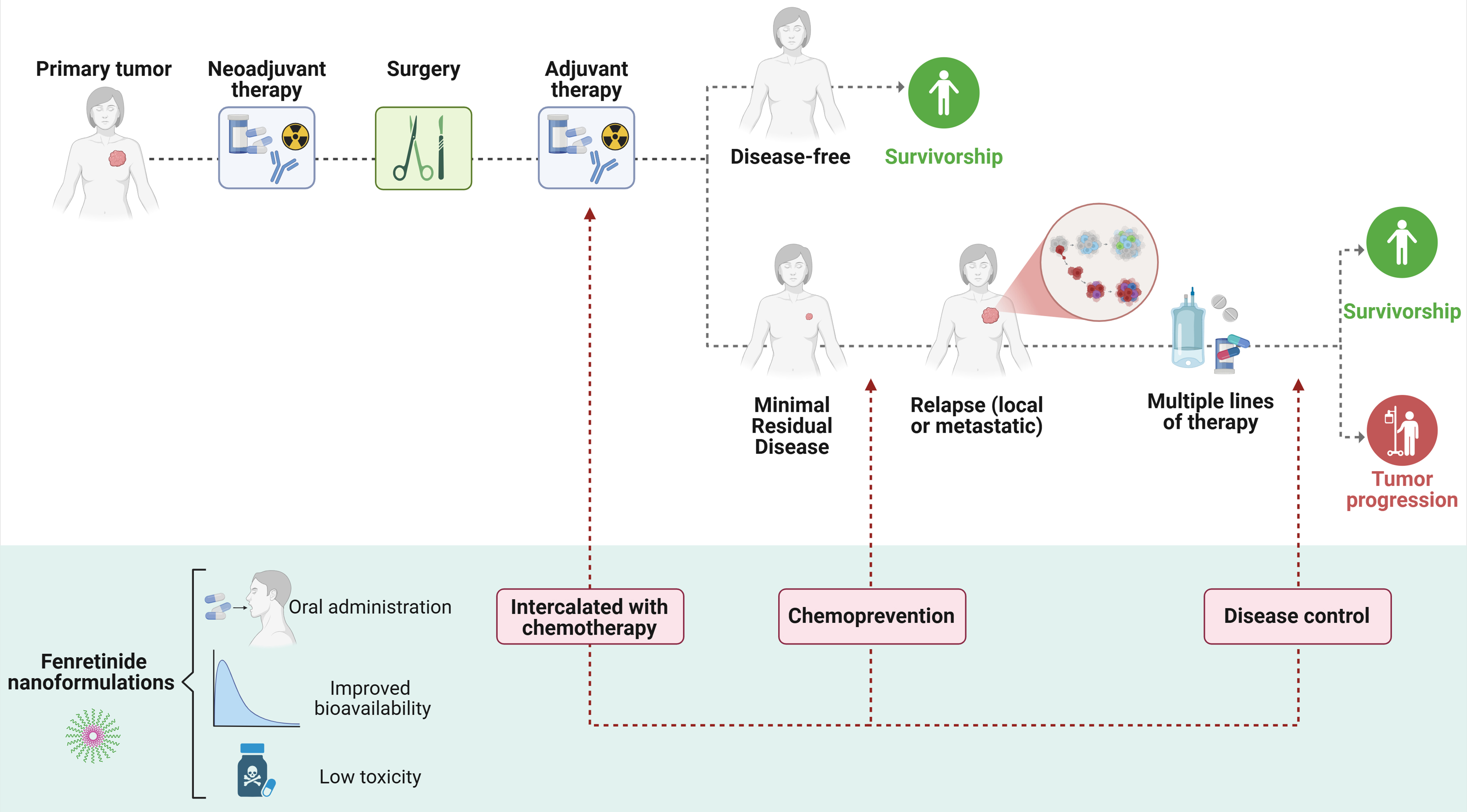
Figure 4. Clinical opportunities for fenretinide in cancer. Schematic overview of possible applications of novel fenretinide nanoformulations in the clinical management of cancer patients. Potential uses of the compound at various stages of patient treatment are suggested: as a chemopreventive agent, as part of standard therapeutic regimens intercalated with chemotherapy agents, or for long-term disease control in advanced tumors. Created in BioRender. Verachi, P. (2026) https://BioRender.com/mu883r2.
DECLARATIONS
Acknowledgments
The graphical abstract was created with BioRender.com [Created in BioRender. Verachi, P. (2026) https://BioRender.com/7fnpzxs].
Authors’ contributions
Conceptualization: Verachi P, Zeuner A
Literature search: Verachi P, Zeuner A, Francescangeli F, De Angelis ML
Design and preparation of figures and tables: Verachi P
Literature review, discussion, writing and editing: Verachi P, Zeuner A, Francescangeli F, De Angelis ML, Dattilo R, Vici A, Landolfo E, Rossi R
Manuscript review: Verachi P, Zeuner A, Francescangeli F, De Angelis ML, Dattilo R, Rossi R
All authors have read and approved the final version of the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Italian Association for Cancer Research (AIRC) Investigator Grants to AZ (AIRC IG 2023 #29148), by Unione Europea - Next Generation EU - PNRR M6C2 - Investimento 2.1 “Valorizzazione e potenziamento della ricerca biomedica del SSN”, PNRRMAD-2022-12376183 (CUP I55E22000570006), and by European Union-Next Generation EU Project M4C2I1.3 “HEAL ITALIA - Health Extended Alliance for Innovative Therapies, Advanced Lab-research, and Integrated Approaches of Precision Medicine”.
Conflicts of interest
Zeuner A is one of the inventors in patent WO2016038534A1 (Fenretinide Complexes). The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Jin Y, Teh SS, Lau HLN, Xiao J, Mah SH. Retinoids as anti-cancer agents and their mechanisms of action. Am J Cancer Res. 2022;12:938-60.
2. Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322-30.
3. Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113:3655-65.
4. Huang M, Ye Y, Chen S, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567-72.
5. Chomienne C, Ballerini P, Balitrand N, et al. All-trans retinoic acid in acute promyelocytic leukemias. II. In vitro studies: structure-function relationship. Blood. 1990;76:1710-7.
7. Hail N Jr, Kim HJ, Lotan R. Mechanisms of fenretinide-induced apoptosis. Apoptosis. 2006;11:1677-94.
8. Orienti I, Francescangeli F, De Angelis ML, et al. A new bioavailable fenretinide formulation with antiproliferative, antimetabolic, and cytotoxic effects on solid tumors. Cell Death Dis. 2019;10:529.
9. Rahmaniyan M, Curley RW Jr, Obeid LM, Hannun YA, Kraveka JM. Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. J Biol Chem. 2011;286:24754-64.
10. Apraiz A, Idkowiak-Baldys J, Nieto-Rementería N, Boyano MD, Hannun YA, Asumendi A. Dihydroceramide accumulation and reactive oxygen species are distinct and nonessential events in 4-HPR-mediated leukemia cell death. Biochem Cell Biol. 2012;90:209-23.
11. Xie H, Zhu F, Huang Z, et al. Identification of mammalian target of rapamycin as a direct target of fenretinide both in vitro and in vivo. Carcinogenesis. 2012;33:1814-21.
12. De Angelis ML, Francescangeli F, Aricò E, et al. A nanoencapsulated oral formulation of fenretinide promotes local and metastatic breast cancer dormancy in HER2/neu transgenic mice. J Exp Clin Cancer Res. 2024;43:296.
13. Fazi B, Bursch W, Fimia GM, et al. Fenretinide induces autophagic cell death in caspase-defective breast cancer cells. Autophagy. 2008;4:435-41.
14. Orienti I, Salvati V, Sette G, et al. A novel oral micellar fenretinide formulation with enhanced bioavailability and antitumour activity against multiple tumours from cancer stem cells. J Exp Clin Cancer Res. 2019;38:373.
15. Du Y, Xia Y, Pan X, et al. Fenretinide targets chronic myeloid leukemia stem/progenitor cells by regulation of redox signaling. Antioxid Redox Signal. 2014;20:1866-80.
16. Liu L, Liu J, Wang H, Zhao H, Du Y. Fenretinide targeting of human colon cancer sphere cells through cell cycle regulation and stress-responsive activities. Oncol Lett. 2018;16:5339-48.
17. Wang H, Zhang Y, Du Y. Ovarian and breast cancer spheres are similar in transcriptomic features and sensitive to fenretinide. Biomed Res Int. 2013;2013:510905.
18. Zhang H, Mi JQ, Fang H, et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proc Natl Acad Sci U S A. 2013;110:5606-11.
19. Sogno I, Venè R, Ferrari N, et al. Angioprevention with fenretinide: targeting angiogenesis in prevention and therapeutic strategies. Crit Rev Oncol Hematol. 2010;75:2-14.
20. Wang D, Pei P, Shea FF, et al. Fenretinide combines perturbation of signaling kinases, cell-extracellular matrix interactions and matrix metalloproteinase activation to inhibit invasion in oral squamous cell carcinoma cells. Carcinogenesis. 2022;43:851-64.
21. Dong R, Gong Y, Meng W, et al. The involvement of M2 macrophage polarization inhibition in fenretinide-mediated chemopreventive effects on colon cancer. Cancer Lett. 2017;388:43-53.
22. Ferrari N, Pfeffer U, Dell’Eva R, Ambrosini C, Noonan DM, Albini A. The transforming growth factor-beta family members bone morphogenetic protein-2 and macrophage inhibitory cytokine-1 as mediators of the antiangiogenic activity of N-(4-hydroxyphenyl)retinamide. Clin Cancer Res. 2005;11:4610-9.
23. GBD 2023 Cancer Collaborators. The global, regional, and national burden of cancer, 1990-2023, with forecasts to 2050: a systematic analysis for the Global Burden of Disease Study 2023. Lancet. 2025;406:1565-86.
24. Francescangeli F, De Angelis ML, Rossi R, et al. Dormancy, stemness, and therapy resistance: interconnected players in cancer evolution. Cancer Metastasis Rev. 2023;42:197-215.
25. D’Alterio C, Scala S, Sozzi G, Roz L, Bertolini G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin Cancer Biol. 2020;60:351-61.
26. Karagiannis GS, Condeelis JS, Oktay MH. Chemotherapy-induced metastasis: molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res. 2019;79:4567-76.
27. Famta P, Shah S, Jain N, et al. Tumor-promoting aftermath post-chemotherapy: a focus on breast cancer. Life Sci. 2022;310:121125.
28. Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501:355-64.
29. Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21:1258-66.
30. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. 2018;20:1349-60.
31. Lüönd F, Tiede S, Christofori G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br J Cancer. 2021;125:164-75.
32. Huang S, Kauffman S. How to escape the cancer attractor: rationale and limitations of multi-target drugs. Semin Cancer Biol. 2013;23:270-8.
33. Doostmohammadi A, Jooya H, Ghorbanian K, Gohari S, Dadashpour M. Potentials and future perspectives of multi-target drugs in cancer treatment: the next generation anti-cancer agents. Cell Commun Signal. 2024;22:228.
35. Delia D, Aiello A, Lombardi L, et al. N-(4-hydroxyphenyl) retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res. 1993;53:6036-41.
36. Mariotti A, Marcora E, Bunone G, et al. N-(4-hydroxyphenyl)retinamide: a potent inducer of apoptosis in human neuroblastoma cells. J Natl Cancer Inst. 1994;86:1245-7.
37. Yan W, Du J, Du Y, et al. Fenretinide targets the side population in myeloma cell line NCI-H929 and potentiates the efficacy of antimyeloma with bortezomib and dexamethasone regimen. Leuk Res. 2016;51:32-40.
38. Veronesi U, Mariani L, Decensi A, et al. Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Ann Oncol. 2006;17:1065-71.
39. Villablanca JG, London WB, Naranjo A, et al. Phase II study of oral capsular 4-hydroxyphenylretinamide (4-HPR/fenretinide) in pediatric patients with refractory or recurrent neuroblastoma: a report from the Children’s Oncology Group. Clin Cancer Res. 2011;17:6858-66.
40. Jasti BR, LoRusso PM, Parchment RE, et al. Phase I clinical trial of fenretinide (NSC374551) in advanced solid tumors. Proc Am Soc Clin Oncol. 2001;20:122a.
41. Potenza RL, Lodeserto P, Orienti I. Fenretinide in cancer and neurological disease: a two-face Janus molecule. Int J Mol Sci. 2022;23:7426.
42. Cooper JP, Reynolds CP, Cho H, Kang MH. Clinical development of fenretinide as an antineoplastic drug: pharmacology perspectives. Exp Biol Med. 2017;242:1178-84.
43. Le Doze F, Debruyne D, Albessard F, Barre L, Defer GL. Pharmacokinetics of all-trans retinoic acid, 13-cis retinoic acid, and fenretinide in plasma and brain of Rat. Drug Metab Dispos. 2000;28:205-8.
44. Alfei S, Zuccari G. Attempts to improve lipophilic drugs’ solubility and bioavailability: a focus on fenretinide. Pharmaceutics. 2024;16:579.
45. Simeone AM, Tari AM. How retinoids regulate breast cancer cell proliferation and apoptosis. Cell Mol Life Sci. 2004;61:1475-84.
46. Lovat PE, Di Sano F, Corazzari M, et al. Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. J Natl Cancer Inst. 2004;96:1288-99.
47. Bassani B, Bartolini D, Pagani A, et al. Fenretinide (4-HPR) targets caspase-9, ERK 1/2 and the Wnt3a/β-catenin pathway in medulloblastoma cells and medulloblastoma cell spheroids. PLoS One. 2016;11:e0154111.
48. Fanjul AN, Delia D, Pierotti MA, Rideout D, Yu JQ, Pfahl M. 4-Hydroxyphenyl retinamide is a highly selective activator of retinoid receptors. J Biol Chem. 1996;271:22441-6.
49. Bu P, Wan YJ. Fenretinide-induced apoptosis of Huh-7 hepatocellular carcinoma is retinoic acid receptor beta dependent. BMC Cancer. 2007;7:236.
50. Yang H, Bushue N, Bu P, Wan YJ. Induction and intracellular localization of Nur77 dictate fenretinide-induced apoptosis of human liver cancer cells. Biochem Pharmacol. 2010;79:948-54.
51. Xiong J, Kuang X, Lu T, et al. Fenretinide-induced apoptosis of acute myeloid leukemia cells via NR4A1 translocation into mitochondria and Bcl-2 transformation. J Cancer. 2019;10:6767-78.
52. Yang H, Zhan Q, Wan YJ. Enrichment of Nur77 mediated by retinoic acid receptor β leads to apoptosis of human hepatocellular carcinoma cells induced by fenretinide and histone deacetylase inhibitors. Hepatology. 2011;53:865-74.
53. Lovat PE, Ranalli M, Annichiarrico-Petruzzelli M, et al. Effector mechanisms of fenretinide-induced apoptosis in neuroblastoma. Exp Cell Res. 2000;260:50-60.
54. Ulukaya E, Pirianov G, Kurt MA, Wood EJ, Mehmet H. Fenretinide induces cytochrome c release, caspase 9 activation and apoptosis in the absence of mitochondrial membrane depolarisation. Cell Death Differ. 2003;10:856-9.
55. Kim YK, Hammerling U. The mitochondrial PKCδ/retinol signal complex exerts real-time control on energy homeostasis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158614.
56. Di Paola M, Cocco T, Lorusso M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry. 2000;39:6660-8.
57. Gentil B, Grimot F, Riva C. Commitment to apoptosis by ceramides depends on mitochondrial respiratory function, cytochrome c release and caspase-3 activation in Hep-G2 cells. Mol Cell Biochem. 2003;254:203-10.
58. Cuperus R, Leen R, Tytgat GA, Caron HN, van Kuilenburg AB. Fenretinide induces mitochondrial ROS and inhibits the mitochondrial respiratory chain in neuroblastoma. Cell Mol Life Sci. 2010;67:807-16.
59. Magaye RR, Savira F, Hua Y, et al. The role of dihydrosphingolipids in disease. Cell Mol Life Sci. 2019;76:1107-34.
60. Lachkar F, Ferré P, Foufelle F, Papaioannou A. Dihydroceramides: their emerging physiological roles and functions in cancer and metabolic diseases. Am J Physiol Endocrinol Metab. 2021;320:E122-30.
61. Erdreich-Epstein A, Tran LB, Bowman NN, et al. Ceramide signaling in fenretinide-induced endothelial cell apoptosis. J Biol Chem. 2002;277:49531-7.
62. Rehman F, Shanmugasundaram P, Schrey MP. Fenretinide stimulates redox-sensitive ceramide production in breast cancer cells: potential role in drug-induced cytotoxicity. Br J Cancer. 2004;91:1821-8.
63. Kraveka JM, Li L, Szulc ZM, et al. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718-28.
64. Zheng W, Kollmeyer J, Symolon H, et al. Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta. 2006;1758:1864-84.
65. Wang H, Maurer BJ, Liu YY, et al. N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Mol Cancer Ther. 2008;7:2967-76.
66. Valsecchi M, Aureli M, Mauri L, et al. Sphingolipidomics of A2780 human ovarian carcinoma cells treated with synthetic retinoids. J Lipid Res. 2010;51:1832-40.
67. Hernández-Tiedra S, Fabriàs G, Dávila D, et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy. 2016;12:2213-29.
68. Tian T, Li X, Zhang J. mTOR Signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int J Mol Sci. 2019;20:755.
69. Benelli R, Monteghirfo S, Venè R, Tosetti F, Ferrari N. The chemopreventive retinoid 4HPR impairs prostate cancer cell migration and invasion by interfering with FAK/AKT/GSK3beta pathway and beta-catenin stability. Mol Cancer. 2010;9:142.
70. Cao J, Xu D, Wang D, et al. ROS-driven Akt dephosphorylation at Ser-473 is involved in 4-HPR-mediated apoptosis in NB4 cells. Free Radic Biol Med. 2009;47:536-47.
71. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461-72.
72. Liu S, Yao S, Yang H, Liu S, Wang Y. Autophagy: regulator of cell death. Cell Death Dis. 2023;14:648.
73. Tiwari M, Bajpai VK, Sahasrabuddhe AA, et al. Inhibition of N-(4-hydroxyphenyl)retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis. 2008;29:600-9.
74. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-7.
75. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-8.
76. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
77. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946-51.
78. Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111-5.
79. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009;26:611-23.
80. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8.
81. Eramo A, Haas TL, De Maria R. Lung cancer stem cells: tools and targets to fight lung cancer. Oncogene. 2010;29:4625-35.
82. Aleksandrova KV, Vorobev ML, Suvorova II. mTOR pathway occupies a central role in the emergence of latent cancer cells. Cell Death Dis. 2024;15:176.
83. Safa AR. Drug and apoptosis resistance in cancer stem cells: a puzzle with many pieces. Cancer Drug Resist. 2022;5:850-72.
84. Jia H, Wei J, Zheng W, Li Z. The dual role of autophagy in cancer stem cells: implications for tumor progression and therapy resistance. J Transl Med. 2025;23:583.
85. Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019;26:690-702.
86. Castaigne S, Chomienne C, Daniel MT, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76:1704-9.
87. Warrell RP Jr, Frankel SR, Miller WH Jr, et al. Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med. 1991;324:1385-93.
88. McKenzie MD, Ghisi M, Oxley EP, et al. Interconversion between tumorigenic and differentiated states in acute myeloid leukemia. Cell Stem Cell. 2019;25:258-72.e9.
89. Carvalho J. Cell reversal from a differentiated to a stem-like state at cancer initiation. Front Oncol. 2020;10:541.
91. Pérez-González A, Bévant K, Blanpain C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat Cancer. 2023;4:1063-82.
92. Farruggia G, Anconelli L, Galassi L, et al. Nano-fenretinide demonstrates remarkable activity in acute promyeloid leukemia cells. Sci Rep. 2024;14:13737.
93. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-3.
94. Villani MG, Appierto V, Cavadini E, et al. 4-oxo-fenretinide, a recently identified fenretinide metabolite, induces marked G2-M cell cycle arrest and apoptosis in fenretinide-sensitive and fenretinide-resistant cell lines. Cancer Res. 2006;66:3238-47.
95. Oki T, Nishimura K, Kitaura J, et al. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0-G1 transition. Sci Rep. 2014;4:4012.
96. Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 2003;63:1684-95.
97. Agudo J, Aguirre-Ghiso JA, Bhatia M, Chodosh LA, Correia AL, Klein CA. Targeting cancer cell dormancy. Nat Rev Cancer. 2024;24:97-104.
98. Balayan V, Guddati AK. Tumor dormancy: biologic and therapeutic implications. World J Oncol. 2022;13:8-19.
99. Hensel JA, Flaig TW, Theodorescu D. Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol. 2013;10:41-51.
100. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834-46.
101. Wang Y, Liu L, Zhang X, Liang T, Bai X. Cancer dormancy and metabolism: from molecular insights to translational opportunities. Cancer Lett. 2025;635:218097.
102. Li X, Ling W, Pennisi A, Khan S, Yaccoby S. Fenretinide inhibits myeloma cell growth, osteoclastogenesis and osteoclast viability. Cancer Lett. 2009;284:175-81.
103. Pienta KJ, Nguyen NM, Lehr JE. Treatment of prostate cancer in the rat with the synthetic retinoid fenretinide. Cancer Res. 1993;53:224-6.
104. Tosetti F, Venè R, Arena G, et al. N-(4-hydroxyphenyl)retinamide inhibits retinoblastoma growth through reactive oxygen species-mediated cell death. Mol Pharmacol. 2003;63:565-73.
105. Ferrari N, Morini M, Pfeffer U, Minghelli S, Noonan DM, Albini A. Inhibition of Kaposi’s sarcoma in vivo by fenretinide. Clin Cancer Res. 2003;9:6020-9.
106. Simile MM, Pagnan G, Pastorino F, et al. Chemopreventive N-(4-hydroxyphenyl)retinamide (fenretinide) targets deregulated NF-κB and Mat1A genes in the early stages of rat liver carcinogenesis. Carcinogenesis. 2005;26:417-27.
107. Reynolds CP, Lemons RS. Retinoid therapy of childhood cancer. Hematol Oncol Clin North Am. 2001;15:867-910.
108. Nieto K, Pei P, Wang D, Mallery SR, Schwendeman SP. In vivo controlled release of fenretinide from long-acting release depots for chemoprevention of oral squamous cell carcinoma recurrence. Int J Pharm. 2018;538:48-56.
109. Garaventa A, Luksch R, Lo Piccolo MS, et al. Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin Cancer Res. 2003;9:2032-9.
110. Liu X, Maurer B, Frgala T, et al. Preclinical toxicology and pharmacokinetics of intravenous lipid emulsion fenretinide. Mol Cancer Ther. 2007;6:C159. Available from: https://aacrjournals.org/mct/article/6/11_Supplement/C159/240578/Preclinical-toxicology-and-pharmacokinetics-of?guestAccessKey=. [Last accessed on 2 Jun 2026].
111. Vaishampayan U, Heilbrun LK, Parchment RE, et al. Phase II trial of fenretinide in advanced renal carcinoma. Invest New Drugs. 2005;23:179-85.
112. Schneider BJ, Worden FP, Gadgeel SM, et al. Phase II trial of fenretinide (NSC 374551) in patients with recurrent small cell lung cancer. Invest New Drugs. 2009;27:571-8.
113. Cheung E, Pinski J, Dorff T, et al. Oral fenretinide in biochemically recurrent prostate cancer: a California cancer consortium phase II trial. Clin Genitourin Cancer. 2009;7:43-50.
114. Moore MM, Stockler M, Lim R, Mok TS, Millward M, Boyer MJ. A phase II study of fenretinide in patients with hormone refractory prostate cancer: a trial of the Cancer Therapeutics Research Group. Cancer Chemother Pharmacol. 2010;66:845-50.
115. Puduvalli VK, Yung WK, Hess KR, et al.; North American Brain Tumor Consortium. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: A North American Brain Tumor Consortium study. J Clin Oncol. 2004;22:4282-9.
116. Colombo N, Formelli F, Cantù MG, et al. A phase I-II preoperative biomarker trial of fenretinide in ascitic ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1914-9.
117. Reynolds CP, Frgala T, Tsao-Wei DD, et al. High plasma levels of fenretinide (4-HPR) were associated with improved outcome in a phase II study of recurrent ovarian cancer: a study by the California Cancer Consortium. J Clin Oncol. 2007;25:5555.
118. Modiano MR, Dalton WS, Lippman SM, Joffe L, Booth AR, Meyskens FL Jr. Phase II study of fenretinide (N-[4-hydroxyphenyl]retinamide) in advanced breast cancer and melanoma. Invest New Drugs. 1990;8:317-9.
119. Maurer BJ, Kalous O, Yesair DW, et al. Improved oral delivery of N-(4-hydroxyphenyl)retinamide with a novel LYM-X-SORB organized lipid complex. Clin Cancer Res. 2007;13:3079-86.
120. Kummar S, Gutierrez ME, Maurer BJ, et al. Phase I trial of fenretinide lym-x-sorb oral powder in adults with solid tumors and lymphomas. Anticancer Res. 2011;31:961-6.
121. Maurer BJ, Kang MH, Villablanca JG, et al. Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: a report from the New Approaches to Neuroblastoma Therapy (NANT) consortium. Pediatr Blood Cancer. 2013;60:1801-8.
122. Maurer BJ, Glade Bender JL, Kang MH, et al. Fenretinide (4-HPR)/Lym-X-Sorb (LXS) oral powder plus ketoconazole in patients with high-risk (HR) recurrent or resistant neuroblastoma: a new approach to Neuroblastoma Therapy (NANT) Consortium trial. J Clin Oncol. 2014;32:10071.
123. Lopez-Barcons L, Maurer BJ, Kang MH, Reynolds CP. P450 inhibitor ketoconazole increased the intratumor drug levels and antitumor activity of fenretinide in human neuroblastoma xenograft models. Int J Cancer. 2017;141:405-13.
124. Thomas JS, El-Khoueiry AB, Maurer BJ, et al. A phase I study of intravenous fenretinide (4-HPR) for patients with malignant solid tumors. Cancer Chemother Pharmacol. 2021;87:525-32.
125. Mohrbacher AM, Yang AS, Groshen S, et al. Phase I study of fenretinide delivered intravenously in patients with relapsed or refractory hematologic malignancies: a California Cancer Consortium trial. Clin Cancer Res. 2017;23:4550-5.
126. Galassi L, Rossi M, Lodeserto P, et al. Naxitamab activity in neuroblastoma cells is enhanced by nanofenretinide and nanospermidine. Pharmaceutics. 2023;15:648.
127. Lodeserto P, Rossi M, Blasi P, Farruggia G, Orienti I. Nanospermidine in combination with nanofenretinide induces cell death in neuroblastoma cell lines. Pharmaceutics. 2022;14:1215.
128. Orienti I, Farruggia G, Nguyen F, et al. Nanomicellar lenalidomide-fenretinide combination suppresses tumor growth in an MYCN amplified neuroblastoma tumor. Int J Nanomedicine. 2020;15:6873-86.
129. Orienti I, Nguyen F, Guan P, et al. A novel nanomicellar combination of fenretinide and lenalidomide shows marked antitumor activity in a neuroblastoma xenograft model. Drug Des Devel Ther. 2019;13:4305-19.
130. Ozpolat B, Tari AM, Mehta K, Lopez-Berestein G. Nuclear retinoid receptors are involved in N-(4-hydroxyphenyl) retinamide (Fenretinide)-induced gene expression and growth inhibition in HL-60 acute myeloid leukemia cells. Leuk Lymphoma. 2004;45:979-85.
131. Nguyen TH, Koneru B, Wei SJ, et al. Fenretinide via NOXA induction, enhanced activity of the BCL-2 inhibitor venetoclax in high BCL-2-expressing neuroblastoma preclinical models. Mol Cancer Ther. 2019;18:2270-82.
132. Fang H, Harned TM, Kalous O, Maldonado V, DeClerck YA, Reynolds CP. Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin Cancer Res. 2011;17:7093-104.
133. Magwere T, Myatt SS, Burchill SA. Manipulation of oxidative stress to induce cell death in Ewing’s sarcoma family of tumours. Eur J Cancer. 2008;44:2276-87.
134. Faes S, Demartines N, Dormond O. Resistance to mTORC1 inhibitors in cancer therapy: from kinase mutations to intratumoral heterogeneity of kinase activity. Oxid Med Cell Longev. 2017;2017:1726078.
135. Roundhill EA, Burchill SA. Detection and characterisation of multi-drug resistance protein 1 (MRP-1) in human mitochondria. Br J Cancer. 2012;106:1224-33.
136. SciTech Development Inc. SciTech development advances to next stage of clinical trial for ST-001 nanoFenretinide. 2025. Available from: https://www.prnewswire.com/news-releases/scitech-development-advances-to-next-stage-of-clinical-trial-for-st-001-nanofenretinide-302348501.html. [Last accessed on 2 Jun 2026].
137. Riggio AI, Varley KE, Welm AL. The lingering mysteries of metastatic recurrence in breast cancer. Br J Cancer. 2021;124:13-26.
138. Rueda OM, Sammut SJ, Seoane JA, et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature. 2019;567:399-404.
139. DeMichele A, Clark AS, Shea E, et al. Targeting dormant tumor cells to prevent recurrent breast cancer: a randomized phase 2 trial. Nat Med. 2025;31:3464-74.
140. Pedersen RN, Esen BÖ, Mellemkjær L, et al. The incidence of breast cancer recurrence 10-32 years after primary diagnosis. J Natl Cancer Inst. 2022;114:391-9.
141. Formelli F, Cleris L. Therapeutic effects of the combination of fenretinide and all-trans-retinoic acid and of the two retinoids with cisplatin in a human ovarian carcinoma xenograft and in a cisplatin-resistant sub-line. Eur J Cancer. 2000;36:2411-9.
142. Kalemkerian GP, Ou X. Activity of fenretinide plus chemotherapeutic agents in small-cell lung cancer cell lines. Cancer Chemother Pharmacol. 1999;43:145-50.
143. Hill DS, Martin S, Armstrong JL, et al. Combining the endoplasmic reticulum stress-inducing agents bortezomib and fenretinide as a novel therapeutic strategy for metastatic melanoma. Clin Cancer Res. 2009;15:1192-8.
144. Cowan AJ, Frayo SL, Press OW, et al. Bortezomib and fenretinide induce synergistic cytotoxicity in mantle cell lymphoma through apoptosis, cell-cycle dysregulation, and IκBα kinase downregulation. Anticancer Drugs. 2015;26:974-83.
145. Pagnan G, Di Paolo D, Carosio R, et al. The combined therapeutic effects of bortezomib and fenretinide on neuroblastoma cells involve endoplasmic reticulum stress response. Clin Cancer Res. 2009;15:1199-209.
146. Liu J, Li J, Zhang JF, Xin XY. Combination of fenretinide and selenite inhibits proliferation and induces apoptosis in ovarian cancer cells. Int J Mol Sci. 2013;14:21790-804.
147. Britt EL, Raman S, Leek K, Sheehy CH, Kim SW, Harada H. Combination of fenretinide and ABT-263 induces apoptosis through NOXA for head and neck squamous cell carcinoma treatment. PLoS One. 2019;14:e0219398.
148. Makena MR, Koneru B, Nguyen TH, Kang MH, Reynolds CP. Reactive oxygen species-mediated synergism of fenretinide and romidepsin in preclinical models of T-cell lymphoid malignancies. Mol Cancer Ther. 2017;16:649-61.
149. Makena MR, Nguyen TH, Koneru B, et al. Vorinostat and fenretinide synergize in preclinical models of T-cell lymphoid malignancies. Anticancer Drugs. 2021;32:34-43.
150. Gopal AK, Pagel JM, Hedin N, Press OW. Fenretinide enhances rituximab-induced cytotoxicity against B-cell lymphoma xenografts through a caspase-dependent mechanism. Blood. 2004;103:3516-20.
151. Cowan AJ, Stevenson PA, Gooley TA, et al. Results of a phase I-II study of fenretinide and rituximab for patients with indolent B-cell lymphoma and mantle cell lymphoma. Br J Haematol. 2017;176:583-90.
152. Decensi A, Robertson C, Guerrieri-Gonzaga A, et al. Randomized double-blind 2 x 2 trial of low-dose tamoxifen and fenretinide for breast cancer prevention in high-risk premenopausal women. J Clin Oncol. 2009;27:3749-56.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.