




New advances in understanding the mechanisms and treatment challenges of ALK-targeted therapy resistance in lung cancer
 0
0 Abstract
Despite the development of various effective anaplastic lymphoma kinase tyrosine kinase inhibitors (ALK-TKIs), therapeutic resistance remains a major challenge. Both on-target and off-target mechanisms have been identified as key contributors to resistance. With the popularization of genetic testing and the development of precision therapies, the prognosis and survival of patients with ALK-positive non-small cell lung cancer (NSCLC) have improved. However, even with second- and third-generation ALK-TKIs, overcoming resistance remains difficult. Resistance frequently arises during approved treatments, underscoring the need for further research to elucidate the molecular events and resistance mechanisms associated with ALK-positive lung cancer. The discovery of anaplastic lymphoma kinase (ALK) rearrangement as an actionable oncogenic driver in NSCLC has established a biomarker-driven treatment paradigm for advanced disease. This article summarizes current knowledge of the mechanisms of resistance to ALK-targeted therapy in lung cancer, including both primary and acquired mechanisms, treatment strategies following resistance, recent therapeutic advances, and the impact of the immune system and tumor microenvironment. A deeper understanding of ALK-targeted therapy resistance is critical for developing new treatment strategies and may provide important insights to guide the diagnosis, treatment, and management of patients with resistant ALK+ lung cancer.
Keywords
INTRODUCTION
Lung cancer remains the leading cause of cancer-related mortality worldwide, with a persistently low five-year overall survival (OS) rate[1]. Non-small cell lung cancer (NSCLC) accounts for about 85% of all lung cancer cases. Targeted therapy has marked a significant advancement in lung cancer treatment and has successfully improved the five-year survival rate of patients with advanced NSCLC[2]. However, the emergence of drug resistance during targeted therapy is common, with resistance to anaplastic lymphoma kinase (ALK) inhibitors being one of the most frequent challenges[3]. Approximately two-thirds of NSCLC patients harbor gene mutations, and the incidence of ALK rearrangements in NSCLC is 3%-7%[4]. ALK can fuse or rearrange with multiple partner genes, among which echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK) is the most common fusion type[5-7].
ALK is regarded as a “diamond target”; however, multiple drug resistance pathways emerge during treatment with ALK tyrosine kinase inhibitors (ALK-TKIs). Even with second- and third-generation ALK-TKIs, overcoming resistance remains a major clinical obstacle[8]. The discovery of ALK rearrangements as operable carcinogenic drivers in NSCLC has fostered a biomarker-oriented treatment paradigm for advanced disease. To date, many ALK-TKIs have been developed and shown to be effective in NSCLC with epidermal growth factor receptor (EGFR) mutations or ALK rearrangements, leading to the approval of highly potent third-generation TKIs such as Osimertinib and Lorlatinib[9,10] [Table 1]. Although these drugs demonstrate remarkable efficacy, resistance remains a major unresolved issue. In patients receiving advanced treatment with Osimertinib or Lorlatinib, resistance mechanisms can be broadly categorized into two types: “on-target” resistance, primarily caused by secondary mutations in the EGFR or ALK kinase domains, and “off-target” resistance, involving alterations in non-target kinases such as bypass pathway activation or phenotypic transformation[29,30].
ALK inhibitors and associated mutations in FDA-approved drugs or ongoing clinical trials
| Drug | Target of mutation | Sensitive mutations | ALK resistance mutations | Regulatory approval | Phase of testing | Ref. |
| 1st generation | ||||||
| Crizotinib (PF-02341066) | ALK rearrangement; ROS1 rearrangement; c-MET | L1198F, E1210K | L1196M, G1269A/S, S1206Y, V1180L, G1202R, C1156Y, I1151Tins, F1174C/L, L1152R/P, L1198P, I1171N/T/S, D1203N | Approved for first-line and subsequent treatment (previously treated patients) | Phase III, completed | [11-15] |
| 2nd generation | ||||||
| Alectinib (RO/CH5424802) | ALK rearrangement; RET; GAK; LTK | C1156Y/T, L1198F, G1269A/S, S1206Y, L1152P/R, F1174C/L/V, I1151Tins | L1196M, V1180L, G1202R, I1171N/T/S, G1269A, S1206Y, F1174L | Approved for first-line and post-crizotinib use | Phase III | [16-18] |
| Ceritinib (LDK378) | ALK rearrangement; ROS1; IGF1R | C1156Y/T, I1171 N/T/S, L1196M, G1269A/S, S1206Y | G1202R, C1156Y, I1151Tins, F1174C/L, L1152R/P | Approved for crizotinib-pretreated patients | Phase III | [19-21] |
| Brigatinib (AP26113) | ALK rearrangement; ROS1; EGFR | C1156Y/T, I1171 N/T/S, L1196M, G1269A, S1206Y, L1198F, L1152R, F1174C/L/V, I1151Tins | S1206C, E1210K, G1202R, E1210K, D1203N | Breakthrough therapy designation for crizotinib-pretreated patients | Phase III | [20-23] |
| 3rd generation | ||||||
| Lorlatinib (PF-06463922) | ALK rearrangement; ROS1 rearrangement | C1156Y/T, I1171N/T/S, L1196M, G1202R, G1269A/S | L1198F, L1256F, G1202R, S1206C/Y, I1171N/T/S, C1156Y, L1196M | EMA: not yet approved; FDA: approval pending | Phase III | [24-26] |
| Ensartinib (X-396) | MET; ROS1; ALK | T1151M, G1269A, L1196M, G1202R | L1198F, G1269A, E1210K, G1202R | EMA: not yet approved; FDA: not yet approved | Phase III | [20,27,28] |
The clinical application of second-generation ALK-TKIs such as alectinib has substantially improved prognosis and survival compared with the chemotherapy era for patients with ALK-positive NSCLC[31]. With the continued progress in precision oncology, molecular testing has become widely adopted in NSCLC management. Since the first report of EML4-ALK rearrangements in NSCLC, numerous additional ALK fusion partners have been identified[32,33]. Consequently, increasing attention has been directed toward the management of ALK-targeted drug resistance in lung cancer. To provide a clearer understanding of its pathogenesis, the role of the immune system and tumor microenvironment, as well as potential therapeutic strategies, this review summarizes current knowledge of the mechanisms and treatment challenges associated with ALK-targeted drug resistance in lung cancer.
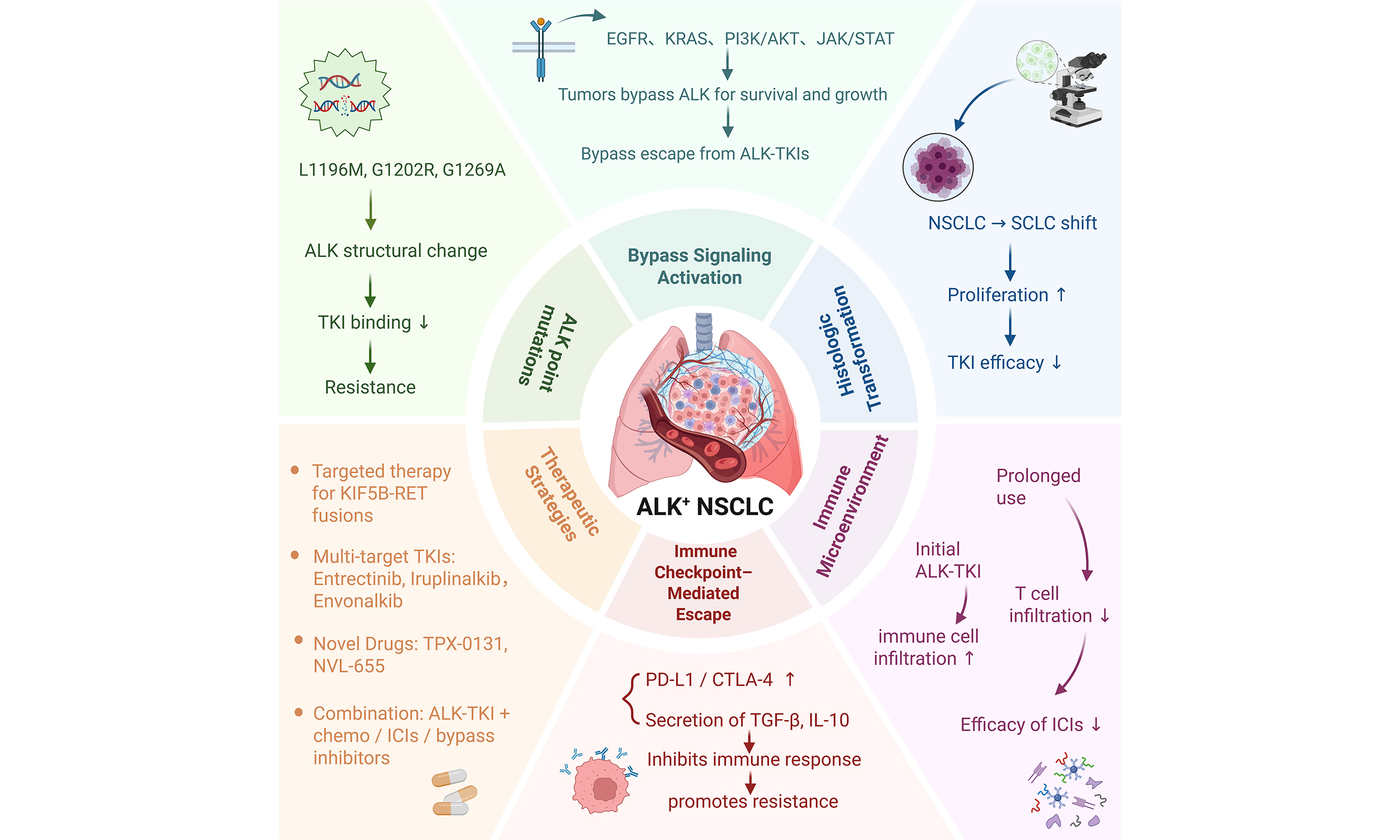
MECHANISM OF ALK-TARGETED DRUG RESISTANCE IN LUNG CANCER
Point mutations in the ALK kinase domain
Point mutations in the ALK gene represent a primary mechanism underlying resistance to ALK-targeted therapies[34]. To date, numerous mutations have been identified within the ALK kinase domain. The most common include: L1196Mlocated in the first peak region of the kinase domain, this “gatekeeper” mutation reduces the binding affinity of ALK inhibitors, thereby conferringdrug resistance[35,36]; G1202R - found in the second peak region of the kinase domain, this mutation increases ATP-binding affinity, which diminishes the effectiveness of drugs[37-39]. S1206Y - also located in the second peak region, this substitution decreases inhibitor binding and promotes resistance; C1156Y/L/F - these variants enhance ALK kinase activity, contributing to resistance against targeted therapy. In addition, several less frequent mutations, such as I1171T/N, F1174C/L/V, R1275Q, and L1152R, also drive therapeutic resistance. Among these, L1196M and G1269A are the most prevalent. Both occur near the ATP-binding pocket and the drug-binding site, directly impairing inhibitor binding[40,41] [Figure 1]. Collectively, these kinase domain mutations disrupt inhibitor binding or enhance kinase activity, representing one of the primary mechanisms of targeted therapy failure. Therefore, comprehensive profiling of such mutations and the development of next-generation inhibitors are critical to extending the therapeutic efficacy of ALK-directed treatment.
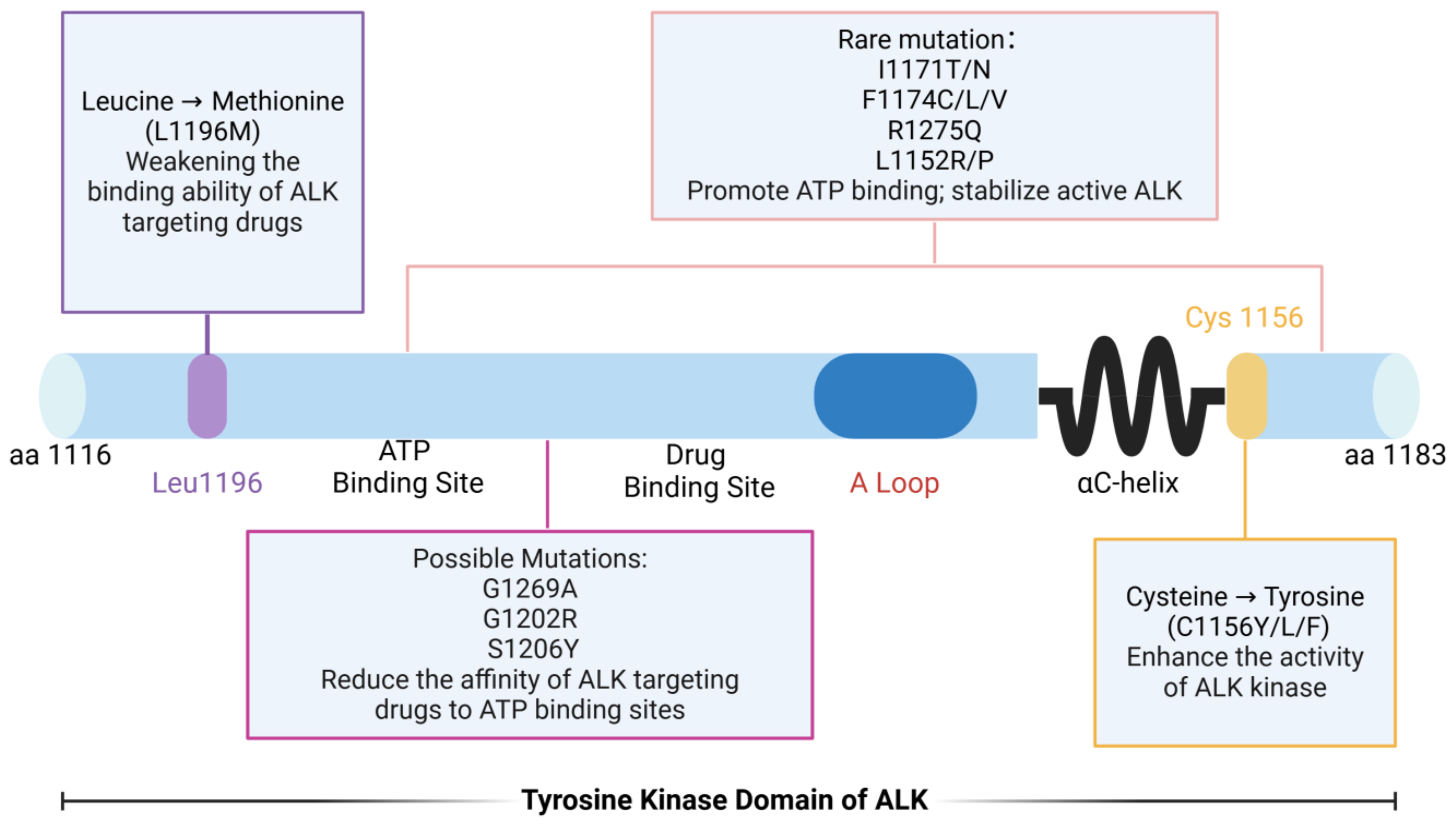
Figure 1. Point mutations within the ALK tyrosine kinase domain and their functional impact. Schematic representation of the ALK tyrosine kinase domain (residues 1116-1183), illustrating the location and functional consequences of representative resistance-associated mutations. The gatekeeper mutation L1196M impairs drug binding. Mutations such as G1202R, S1206Y, and G1269A reduce inhibitor affinity at the ATP-binding pocket. Variants including I1171T/N, F1174C/L/V, R1275Q, and L1152R/P increase ATP binding and stabilize the active kinase conformation. Substitutions at C1156 (C1156Y/L/F) enhance ALK kinase activity, further promoting resistance. Created in BioRender. Long, M. (2025). https://BioRender.com/4hhix6x. ALK: Anaplastic lymphoma kinase; ATP: adenosine triphosphate.
Bypass signaling pathway activation and resistance to ALK-targeted therapy
ALK is a receptor tyrosine kinase whose aberrant activation in lung cancer promotes cell proliferation, survival, and metastasis. ALK-targeted therapies, including agents such as Crizotinib and Alectinib, have been extensively employed in the treatment of ALK-positive NSCLC[42] [Table 1]. However, due to the complexity of intracellular signaling networks, tumor cells frequently develop resistance, leading to the activation of alternative pathways that bypass ALK inhibition. The main bypass signaling pathways implicated in ALK-targeted therapy resistance are outlined below [Figure 2].
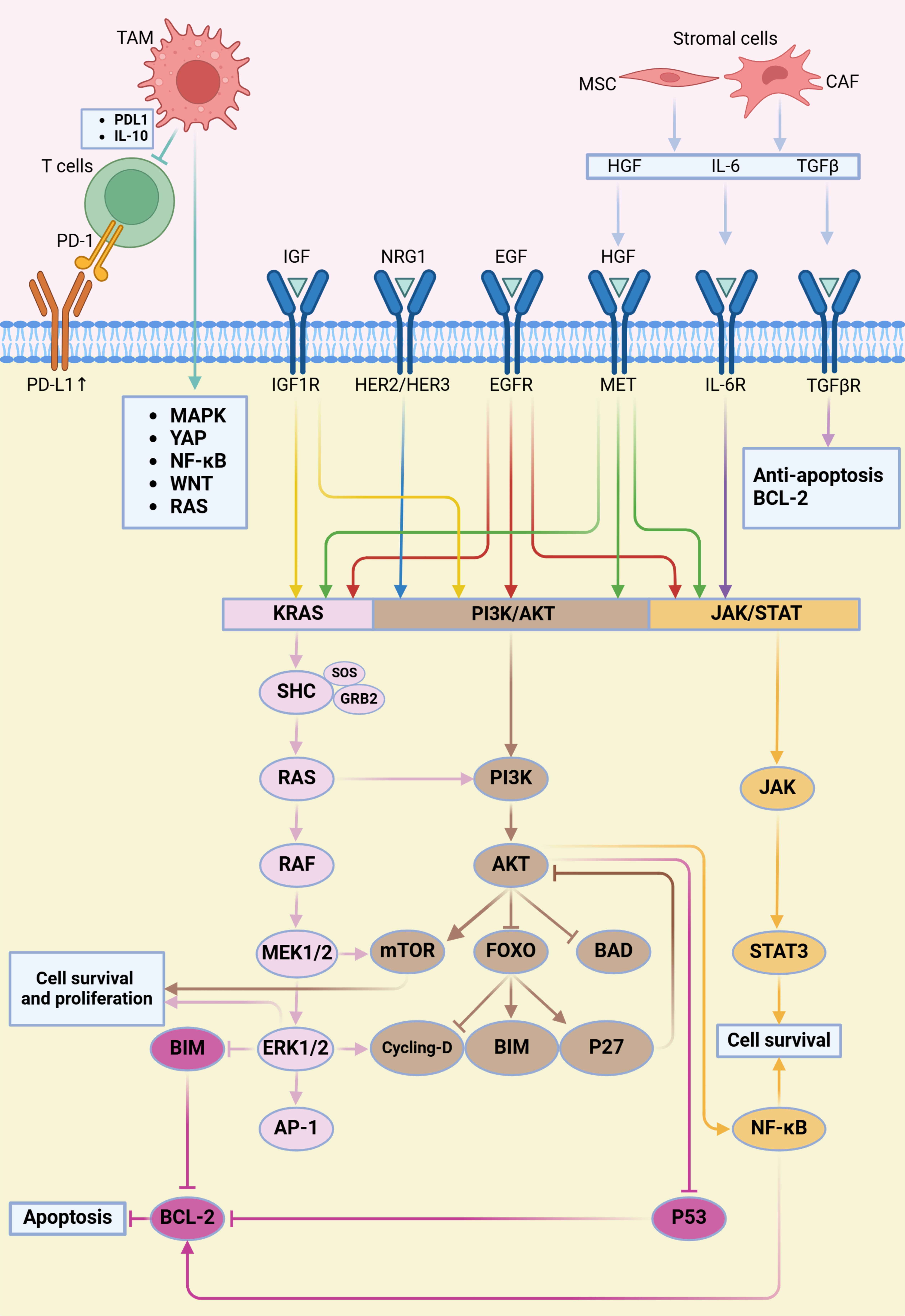
Figure 2. ALK-TKI resistance mediated by the tumor microenvironment and bypass signaling activation. This figure illustrates how the tumor microenvironment promotes resistance to ALK-TKIs through activation of bypass signaling pathways. Stromal components such as CAFs and MSCs secrete cytokines and growth factors including IL-6, TGFβ, and HGF. Together with ligands such as IGF, NRG1, and EGF, these molecules activate corresponding receptors on tumor cells. Receptor activation triggers downstream signaling pathways including RAS–RAF–MEK–ERK, PI3K–AKT, and JAK–STAT, ultimately enhancing cell survival and inhibiting apoptosis through mediators such as BCL-2 and NF-κB. This process reduces the effectiveness of ALK inhibitors. Additionally, TAMs contribute to immune evasion by secreting IL-10 and inducing PD-L1 expression on tumor cells, thereby suppressing T cell-mediated antitumor immunity. Both TAMs and T cells further modulate oncogenic signaling by releasing factors that activate the MAPK, YAP1, NF-κB, WNT, and RAS pathways. Created in BioRender. Long, M. (2025). https://BioRender.com/4cy0tmf. ALK-TKI: Anaplastic lymphoma kinase tyrosine kinase inhibitor; CAFs: cancer-associated fibroblasts; MSCs: mesenchymal stem cells; PI3K: phosphatidylinositol 3-kinase; JAK: Janus kinase; STAT: signal transducer and activator of transcription; NF-κB: nuclear factor-κB; TAMs: tumor-associated macrophages; PD-L1: programmed death ligand 1; YAP1: Hippo-Yes-associated protein 1.
EGFR-mediated bypass activation
The EGFR is a receptor tyrosine kinase that interacts closely with ALK signaling. In ALK-positive NSCLC, EGFR mutations and overexpression have been associated with resistance to ALK inhibitors[43]. EGFR activation stimulates downstream signaling molecules independently of ALK, thereby promoting resistance to therapy[44]. Among bypass mechanisms, EGFR activation is one of the most frequent, particularly after treatment with first- and second-generation ALK inhibitors such as Crizotinib, Ceritinib, and Alectinib. At the molecular level, increased EGFR autophosphorylation and upregulation of EGFR ligands reactivate downstream cascades such as the phosphatidylinositol 3-kinase (PI3K)/AKT and MAPK/ERK pathways, enabling tumor cells to reduce dependence on ALK signaling[41]. Notably, heparin-binding EGF-like growth factor (HB-EGF) has been reported to induce crizotinib resistance by activating EGFR and stimulating the ERK1/2 and AKT pathways[45]. These findings highlight the critical role of EGFR-mediated bypass signaling in limiting the long-term efficacy of ALK inhibitors.
KRAS-mediated bypass activation
Kirsten rat sarcoma viral oncogene homolog (KRAS) is a small GTPase that regulates cell growth and survival. KRAS mutations contribute to ALK inhibitor resistance in ALK-positive NSCLC by activating downstream signaling pathways, including MAPK/ERK and PI3K/AKT. These mutations are linked to poor prognosis and higher recurrence risk[46,47]. In addition, recent studies have shown that in BRAF-mutant melanoma, aberrant ALK activation can restore MAPK pathway signaling through bypass mechanisms, conferring resistance to BRAF inhibitors (BRAFi)[48]. Overexpression or fusion of ALK can reinstate ERK phosphorylation and sustain tumor proliferation, suggesting that ALK inhibitors may help overcome BRAFi resistance. Conversely, although direct evidence in NSCLC is limited, MAPK pathway reactivation - including BRAF and MEK/ERK signaling - has been observed in ALK inhibitor resistance models, potentially reducing the efficacy of agents such as Ceritinib and Alectinib[49,50]. Collectively, BRAF and KRAS mutations appear to converge with ALK signaling on shared downstream pathways, driving resistance phenotypes. Given its clinical significance, aberrant KRAS pathway activation in NSCLC has become an active focus of therapeutic research[51].
PI3K/AKT-mediated bypass activation
The PI3K/AKT pathway is essential for cell survival and proliferation[52]. Hyperactivation of this pathway enhances proliferation, survival, and metastasis in lung cancer, and has been implicated in resistance to ALK inhibitors[53,54]. In ALK-TKI resistance, PI3K/AKT activation often occurs independently of ALK signaling, supporting tumor growth by suppressing pro-apoptotic proteins and amplifying anti-apoptotic signals. Clinical and preclinical studies have demonstrated that the PI3K/AKT pathway is reactivated in ALK-positive NSCLC after Crizotinib and other TKI treatments, often through bypass activation involving EGFR or IGF-1R. Notably, secondary EGFR mutations that trigger PI3K/AKT reactivation have been linked to crizotinib resistance and diminished therapeutic responses[55]. These findings underscore the importance of simultaneously targeting ALK and key downstream effectors such as PI3K/AKT to overcome acquired resistance in ALK-positive NSCLC.
JAK/STAT-mediated bypass activation
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway regulates cell growth and differentiation. In ALK-positive NSCLC, aberrant activation of the JAK/STAT pathway has been correlated with resistance to ALK inhibitors[56]. This signaling cascade promotes tumor proliferation by downregulating cell cycle inhibitors and upregulating cell cycle-promoting molecules[57].
Beyond these well-established pathways, others - including the Wnt/β-catenin and NF-κB pathways - may also participate in bypass-mediated resistance to ALK-targeted therapy[58,59]. Their aberrant activation further compromises the effectiveness of ALK inhibitors. Consequently, overcoming bypass resistance requires a deeper mechanistic understanding of signaling networks in lung cancer, which will inform the development of novel therapeutic strategies and improve the long-term efficacy of ALK-targeted treatments.
Small cell transformation in the progression of ALK-targeted drug resistance in lung cancer
Small cell lung cancer (SCLC) is a highly malignant neuroendocrine subtype of lung cancer with a poor prognosis. It is characterized by aggressive behavior, rapid tumor growth, and widespread metastases[60]. Transformation into SCLC represents a resistance mechanism through which NSCLC can evade immunotherapy, chemotherapy, and targeted therapy. In a subset of patients with ALK-positive NSCLC, histologic transformation to SCLC may occur, resulting in substantial resistance to ALK-targeted therapy. The 5-year survival rate for SCLC remains extremely low, at approximately 5%-10%, compared with NSCLC, which generally exhibits slower growth and delayed invasion or metastasis[61]. Transformed SCLC is considered a distinct phenotype from de novo SCLC, although the two share certain molecular, histopathological, and clinical features, as well as similar patterns of drug responsiveness. Nonetheless, de novo SCLC and transformed SCLC differ in their underlying pathogenesis and tumor microenvironment [Figure 3]. In ALK-positive NSCLC, SCLC transformation typically emerges after exposure to ALK inhibitors, signifying advanced drug resistance. This process may be associated with reduced expression of the ALK fusion gene, dysregulation of cell proliferation signaling pathways, and altered transcription factor activity[62].
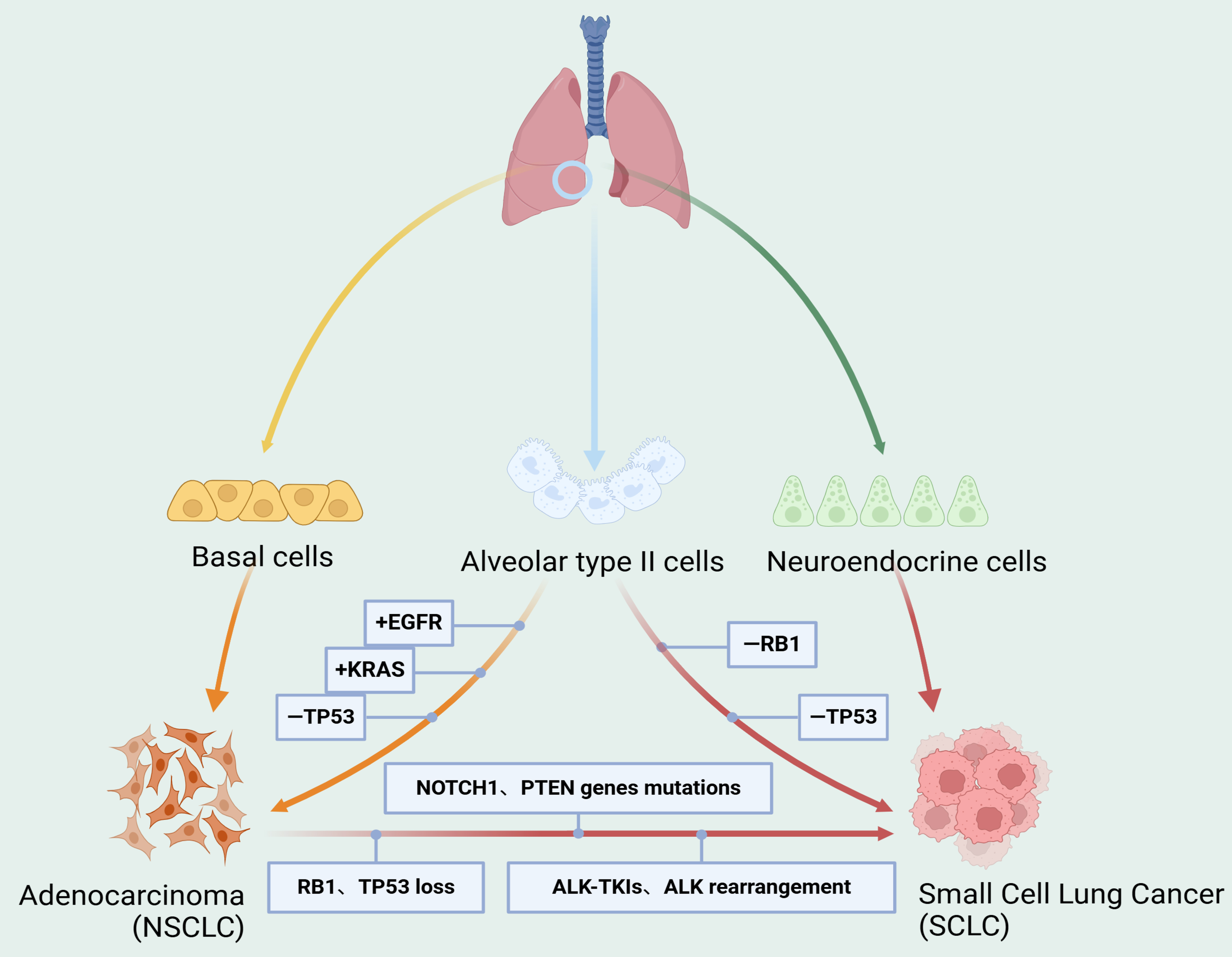
Figure 3. Mechanisms of small cell transformation in the progression of ALK-targeted resistance in lung cancer. SCLC can originate from neuroendocrine cells, while adenocarcinoma typically arises from basal cells. Alveolar type II cells may serve as a potential origin for both SCLC and adenocarcinoma. During SCLC transformation, multiple genetic alterations and signaling pathway activations are involved, including RB1 and TP53 loss, mutations in genes such as NOTCH1 and PTEN, ALK-TKI resistance, and ALK rearrangements. Created in BioRender. Long, M. (2025). https://BioRender.com/ogqzb3a. SCLC: Small cell lung cancer; ALK-TKI: anaplastic lymphoma kinase tyrosine kinase inhibitor.
For NSCLC patients who undergo SCLC transformation, timely confirmation of diagnosis and initiation of individualized treatment are critical. To overcome this resistance mechanism, further investigation into the molecular basis of transformation and the development of corresponding treatment strategies are urgently needed. Currently, no clear or standardized treatment strategy exists for ALK-positive NSCLC patients who develop SCLC transformation. Early studies suggest that traditional SCLC treatments, such as chemotherapy and radiotherapy, may provide some benefit in this setting[63,64]. In addition, novel therapeutic drugs targeting SCLC are under development, though no consensus standard of care is available for patients who experience transformation after ALK-TKI therapy. Re-biopsy at the time of acquired resistance has been recommended to guide alternative treatment selection[65]. For instance, Cisplatin combined with Irinotecan (the standard SCLC regimen in Japan) has achieved sustained partial remission of primary tumors as well as partial remission at metastatic sites[66]. These findings underscore the need to promptly adjust and individualize treatment plans for patients with SCLC transformation to maximize therapeutic outcomes.
Preliminary studies have reported clinical outcomes in patients with ALK-positive NSCLC who develop SCLC transformation. A systematic review analyzing 17 cases of lung adenocarcinoma and 16 cases treated with first- or second-generation TKIs found that patients had a median OS of 6 months following SCLC transformation, whereas those treated solely with Osimertinib had a median OS of just 2 months[67]. Another study of 58 patients with NSCLC reported a median time to transformation of 17.8 months, a median OS of 31.5 months from initial diagnosis, and a median survival of 10.9 months following transformation[68]. Biopsy remains the most reliable method for confirming SCLC transformation. Conventional SCLC regimens, including chemotherapy and radiotherapy, may offer limited efficacy in these cases[69]. However, several challenges persist. On one hand, conventional treatments are often associated with substantial toxicity, which may significantly affect quality of life. On the other hand, novel therapies for SCLC remain in the early stages of development, with their long-term safety and efficacy yet to be established[70,71]. Therefore, when managing ALK-positive NSCLC patients who experience SCLC transformation, it is essential to consider the patient’s overall condition, prior treatment history, and quality of life in tailoring an appropriate therapeutic plan. Meanwhile, further research into the mechanisms of SCLC transformation is needed to identify novel therapeutic targets and strategies, ultimately expanding treatment options and improving outcomes for these patients.
THE RELATIONSHIP BETWEEN ALK-TARGETED THERAPY AND THE IMMUNE MICROENVIRONMENT
Although immune checkpoint inhibitors (ICIs) represent a key treatment option for advanced NSCLC, patients with ALK-rearranged NSCLC generally fail to derive significant clinical benefit[72,73]. This lack of response may be attributed to the unique features of the TME[74]. Research indicates that the immune microenvironment plays a critical role in the development of resistance to ALK-targeted therapy. Initially, ALK inhibitors can enhance immune cell infiltration and activation, promoting cancer cell apoptosis. However, prolonged treatment may gradually alter the TME, resulting in reduced immune cell infiltration and activation, allowing lung cancer cells to escape immune surveillance and develop therapeutic resistance[75]. Moreover, ALK-targeted therapy can modulate the expression and activity of immune checkpoint molecules, including CTLA-4, programmed death ligand 1 (PD-L1), and PD-1, which suppress immune cell function and further facilitate immune escape. To date, no survival analyses have been conducted based on PD-L1 expression in ALK-positive patients receiving TKI therapy. Instead, existing studies have primarily assessed the immune landscape using markers such as PD-L1, PD-1, CD3, and CD8[76-78]. Therefore, therapeutic strategies aimed at enhancing immune cell activity - such as combining ICIs with ALK-targeted agents - may help overcome resistance and improve outcomes in lung cancer patients.
The advent of ALK-targeted therapy has significantly improved survival rates in lung cancer. Nevertheless, during treatment, patients often develop local and systemic immune dysregulation, especially when resistance to ALK inhibitors emerges. In resistant cases, both the number and composition of immune cells within the TME - including B cells, T cells, and natural killer (NK) cells - are altered. These immune cells can interact with tumor cells, driving further progression and metastasis[79]. Importantly, ALK-targeted therapies can reshape the immune microenvironment in ALK fusion-positive NSCLC. Such therapies may increase immune cell infiltration while suppressing the expression of immunosuppressive molecules, thereby enhancing the immune system’s ability to recognize and eliminate tumor cells. This effect could improve patient responsiveness to immunotherapy[80]. Taken together, understanding the complex interactions between ALK-targeted therapy and the immune microenvironment is crucial for developing more effective treatment approaches and improving clinical outcomes in patients with lung cancer.
ALK-TARGETED THERAPY RESISTANCE IN LUNG CANCER AND IMMUNE ESCAPE RELATED TO IMMUNE CHECKPOINTS
Resistance to ALK-targeted therapy in lung cancer is also related to immune escape mechanisms involving immune checkpoints. Studies have shown that PD-L1 expression is significantly elevated in patients with ALK-targeted therapy resistance, which may enable tumor cells to evade immune detection and elimination[81,82]. Consequently, ICIs may enhance treatment outcomes in patients with ALK-resistant lung cancer. Immune escape contributes to the development of resistance to ALK-targeted therapies by allowing tumor cells to avoid recognition and clearance by the immune system through multiple mechanisms[83]. In lung cancers resistant to ALK-targeted drugs, tumor cells may achieve immune escape via several pathways: First, they may downregulate the expression of tumor antigens, reducing immune system recognition and attack[84]. Second, they may activate immunosuppressive pathways, including the PD-L1/PD-1 and CTLA-4 axes, thereby suppressing immune cell activation. Additionally, tumor cells may secrete immunosuppressive factors such as TGF-β and IL-10, which induce immune tolerance and further prevent immune-mediated attack. Upregulation of immune checkpoint molecules such as PD-L1 represents a key mechanism by which tumor cells evade immune surveillance following resistance to ALK-targeted therapy.
Current therapeutic strategies to overcome immune escape in ALK-targeted therapy-resistant lung cancer mainly involve immunotherapy and combination therapy. Immunotherapy aims to inhibit immunosuppressive pathways or enhance immune cell activation to strengthen the immune system’s ability to attack tumor cells. Combination therapy, on the other hand, employs multiple treatment modalities, such as targeted therapy, radiotherapy, chemotherapy, and immunotherapy, simultaneously to attack tumor cells from different angles, thereby improving treatment efficacy and tolerability[85]. Addressing immune escape is a crucial aspect of managing ALK-targeted therapy resistance, and treatment strategies must consider tumor characteristics, patient condition, therapeutic efficacy, and safety.
METHODS FOR TREATING ALK-TARGETED THERAPY-RESISTANT LUNG CANCER
Lung cancers harboring the ALK fusion gene can often be controlled with ALK-targeted inhibitors, which help prevent disease progression. However, long-term use of these drugs can lead to resistance, posing a significant challenge in clinical management. Developing new strategies to treat ALK-targeted therapy-resistant lung cancer is therefore crucial[86,87]. Currently, several approaches have been explored to overcome ALK resistance [Table 2], including the following:
Summary of alternative strategies for the treatment of targeted drug resistance in ALK
| Drug regimen | Study phase | Results | Clinical trials/ref. |
| KIF5B-RET inhibitor (LOXO-292) | I | Ongoing | NCT03157128 |
| Immunotherapy (PD-L1 inhibitors) | Preclinical/clinical | Upregulation of PD-L1. Demonstrated marked antitumor efficacy compared with monotherapy in preclinical ALK-positive NSCLC models. In clinical trials, combinations showed some activity but with severe adverse events | [85,88-91] |
| Targeting drug-tolerant Persister cells | IV | Local consolidative approaches used to eliminate persister cells in areas of residual disease | NCT02314364 [92] |
| Multitarget therapy | |||
| Alectinib + Bevacizumab (anti-VEGF mAb) | I/II | Upfront ALK and VEGFR inhibition re-sensitized ALK-TKI-resistant ALK-positive NSCLC cell lines. Lorlatinib plus bevacizumab achieved disease regression | NCT02521051 [93,94] |
| Ceritinib + Everolimus (mTOR inhibitor) | I/Ib | Combination of ALK and mTOR inhibition significantly reduced the proliferation of ALK-TKI-resistant cell lines | NCT02321501 [95-98] |
| Ceritinib + Trametinib (MEK inhibitor) | Preclinical/clinical | Enhanced the magnitude and duration of initial drug response in untreated ALK-positive NSCLC cell lines and overcame resistance in ALK-TKI-resistant lines | NCT03087448 NCT03202940 [99-104] |
| Crizotinib + Dacomitinib (HER2 inhibitor) | I | Excessive toxicity | NCT01121575 [96] |
| Lorlatinib + Crizotinib | I/II | Unknown | NCT04292119 [105] |
| Lorlatinib + TNO155 | I/II | Unknown | NCT04292119 [106] |
| EGFR inhibitors | Preclinical | Improved therapeutic efficacy compared with ALK-TKI monotherapy | [107-111] |
| New ALK-targeting drugs | |||
| TPX-0131 | I | Terminated (adverse change in risk-benefit ratio) | NCT04849273 |
| NVL-655 (Neladalkib) | I/II | Ongoing | NCT05384626 |
| WX-0593 | II | Ongoing (recruiting) | NCT04641754 |
| TQ-B3139 | II | Unknown | NCT04056572 |
| Combination therapy | |||
| Chemotherapy (Irinotecan) + Alectinib | / | Partial response in primary lesion and metastases | [65] |
| Immunotherapy (Nivolumab) + Ceritinib | Ib | Demonstrated activity | [91] |
| Ipilimumab (CTLA-4 inhibitor) + Nivolumab (PD-1 inhibitor) | / | Improved response rates in advanced NSCLC | [106] |
KIF5B-RET targeted therapy
KIF5B-RET fusion genes are more frequently observed in ALK-negative lung cancers, accounting for approximately 5%-10% of cases. This fusion gene leads to a unique tumor biological phenotype that is highly sensitive to RET-targeted drugs, offering a promising therapeutic option. Several RET inhibitors have demonstrated encouraging efficacy in clinical trials. Therefore, KIF5B-RET targeted therapy presents a potential strategy for patients with ALK-resistant lung cancer[112,113]. RET-targeted drugs mainly include polypeptide amides, pyrrolothiazoles, triazoles, and related compounds. These agents act by directly or indirectly inhibiting RET kinase activity, thereby blocking downstream signaling pathways and suppressing tumor growth. Clinical trials have demonstrated excellent efficacy, and some of these drugs have received FDA approval for treating KIF5B-RET-positive lung cancer[114].
Beyond selective RET inhibition, research has increasingly focused on developing dual RET/ALK inhibitors, which may provide clinical advantages in tumors with complex resistance mechanisms or concurrent oncogenic alterations. Notably, Song et al. identified benzo[b]kazolone, a compound initially designed as an ALK inhibitor, which also exhibits potent activity against both wild-type and mutant forms of ALK (including L1196M and C1156Y), as well as mutant RET[115]. This compound demonstrated low nanomolar IC50 values across multiple kinase targets, indicating a broad-spectrum inhibition profile. In vivo xenograft studies further confirmed its robust antitumor efficacy, supporting its potential application in NSCLC cases driven by RET fusions or those with acquired ALK inhibitor resistance. These findings suggest that dual RET/ALK inhibition may represent a promising strategy to overcome limitations of single-agent kinase inhibitors[116].
It is important to note that KIF5B-RET targeted therapy is not suitable for all lung cancer patients; genetic testing is necessary to identify candidates. Additionally, targeted therapies may cause side effects and can still encounter resistance, requiring careful evaluation and management by healthcare providers[117]. In summary, KIF5B-RET targeted therapy offers a novel approach that offers renewed hope for patients with ALK-targeted therapy-resistant lung cancer. Accurate molecular subtyping and target screening are critical to enabling personalized treatment strategies for patients with acquired resistance.
Immunotherapy
Immunotherapy activates the patient’s immune system to recognize and eliminate cancer cells. Although it is not yet widely established for the treatment of ALK-targeted therapy-resistant lung cancer, some studies suggest that immunotherapy may offer benefits in ALK-positive NSCLC[118,119]. In recent years, ICIs (such as PD-1 and PD-L1 inhibitors) have shown efficacy in lung cancer treatment, including ALK-positive cases[120,121] [Figure 4]. Evidence also indicates that ALK inhibitors can enhance the effects of immunotherapy, making combination regimens a potential strategy for patients with ALK-targeted therapy resistance. Several clinical trials are currently exploring this approach. The Phase III KEYNOTE-789 trial (NCT03515837) is evaluating chemotherapy with or without Pembrolizumab in NSCLC patients, while the Phase III ATLAS trial (NCT03991403) is assessing the combination of atezolizumab and bevacizumab with chemotherapy in patients harboring EGFR mutations or ALK rearrangements[29]. Crizotinib, a first-line ALK inhibitor for advanced ALK-positive NSCLC, demonstrates initial efficacy; however, disease progression is inevitable. In contrast, immunotherapy approaches, including PD-1 inhibitors such as Nivolumab, have produced durable clinical responses and contributed to prolonged OS in some lung cancer patients[85]. Notably, results from the ALK/EGFR-positive subgroup of the IMpower150 trial demonstrated that combination therapy significantly improved patient survival, suggesting that this approach may partially overcome the immune tolerance associated with ALK-positive tumors[122]. Moreover, novel PD-1/VEGF bispecific antibodies, such as AK112, have shown promising efficacy in ongoing clinical trials, offering new perspectives for future combination strategies[123]. Currently, several ICIs, including Nivolumab, Atezolizumab, Durvalumab, and Pembrolizumab, have received regulatory approval for the treatment of lung cancer. The introduction of these agents has transformed the therapeutic landscape of advanced NSCLC, markedly improving outcomes in certain patient populations[124]. Although ICI monotherapy has shown limited efficacy in ALK-positive NSCLC, combining ICIs with chemotherapy, anti-angiogenic therapy, or second-/third-generation ALK-TKIs may represent a promising direction to overcome therapeutic resistance and provide renewed hope for affected patients.
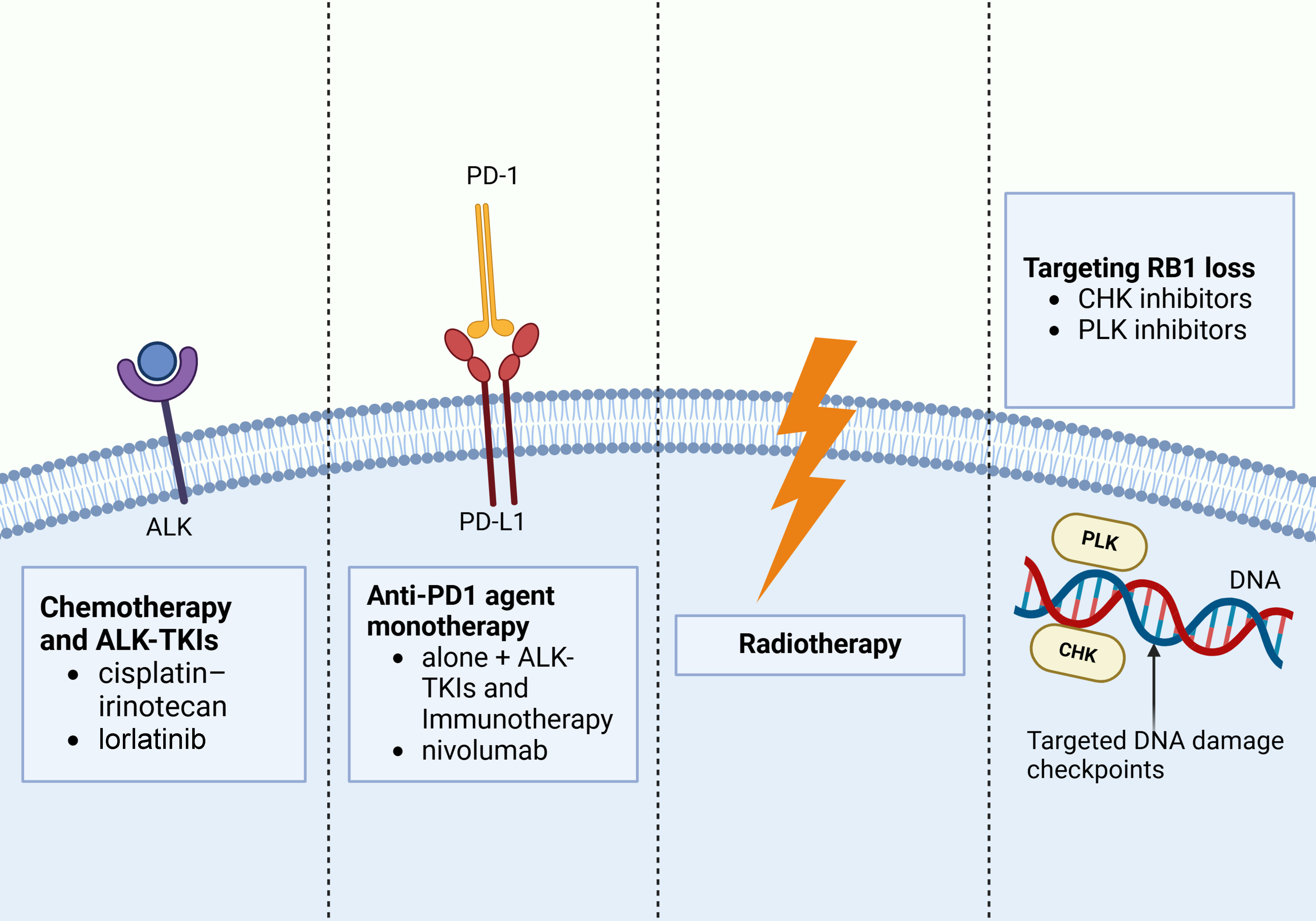
Figure 4. Potential therapeutic targets for transformed SCLC from ALK-mutant NSCLC. These treatments include chemotherapy, ALK-TKIs, immunotherapy, radiation therapy, and CHK and PLK inhibitors targeting RB1 loss. Created in BioRender. Long, M. (2025). https://BioRender.com/gtgq662. SCLC: Small cell lung cancer; ALK: anaplastic lymphoma kinase; NSCLC: non-small cell lung cancer; TKIs: tyrosine kinase inhibitors; CHK: checkpoint kinase; PLK: polo-like kinase.
Multitarget therapy
In addition to ALK gene fusions, ALK-positive lung cancer may also harbor other molecular alterations that contribute to tumor growth and progression. Therefore, simultaneously targeting multiple molecules may be more effective in controlling disease progression in cases with acquired resistance to ALK-targeted therapy. Recent evidence suggests that dual targeting of ALK and EGFR or HER2 can provide therapeutic benefits in ALK inhibitor-resistant lung cancer[125,126]. For example, combining ALK inhibitors with EGFR inhibitors (such as Gefitinib) suppresses both ALK and EGFR signaling pathways[127,128]. In transgenic mouse models carrying EGFR mutations (Del19-T790M and L858R-T790M), the combination of Crizotinib and the third-generation EGFR inhibitor WZ4002 exhibited potent antitumor activity[129]. Similarly, in NCI-H3122 cells with hyperactivated EGFR signaling, resistance to Ceritinib and Alectinib was effectively overcome by co-treatment with the EGFR inhibitor Afatinib[130]. Osimertinib, a third-generation EGFR TKI that irreversibly binds its target, has been established as the standard first-line treatment for NSCLC patients with EGFR mutations. In the FLAURA trial, Osimertinib achieved superior OS and progression-free survival (PFS) compared with first-generation EGFR TKIs such as Erlotinib and Gefitinib. The subsequent FLAURA-2 study further indicated that combining chemotherapy with Osimertinib extended PFS from 16.7 to 25.5 months, highlighting the potential synergistic benefits of multitargeted strategies in specific oncogenic contexts[131,132].
Several novel multitarget TKIs have also demonstrated promising clinical potential. Entrectinib, a broad-spectrum inhibitor of tropomyosin receptor kinase (TRK), ROS1, and ALK, has shown efficacy in tumors with diverse gene fusions and is under evaluation in the Phase II STARTRK-2 clinical trial[133]. Iruplinalkib (WX-0593) and Envonalkib (TQ-B3139), which selectively target ROS1, ALK, and MET, have received approval in China to treat locally advanced or metastatic ALK-positive NSCLC in patients who progressed on or were intolerant to Crizotinib[134,135]. Collectively, these findings suggest that multitargeted approaches may help overcome resistance to ALK inhibitors and improve clinical outcomes. Systematic evaluation of resistance mechanisms and dynamic adjustment of therapeutic regimens will be essential to optimize long-term survival.
Novel ALK-targeted drugs
Several novel ALK-targeted drugs have been developed and are currently being tested in clinical studies. These next-generation inhibitors suppress ALK protein activation through different mechanisms, thereby helping to overcome resistance to earlier ALK inhibitors. For instance, Lorlatinib, a third-generation ALK inhibitor approved by the FDA for the treatment of ALK-positive NSCLC, effectively inhibits a wide range of ALK variants, including those that have acquired resistance to prior ALK-TKIs[136]. It has demonstrated strong activity against multiple acquired ALK mutations, such as G1202R, and can restore sensitivity in patients resistant to Crizotinib. Lorlatinib also shows potent inhibitory activity against ALK and ROS1 mutations and resistance mechanisms[137,138]. However, with continued use, new compound mutations may arise, ultimately leading to resistance to Lorlatinib itself.
To address drug-resistant mutations caused by long-term ALK-TKI therapy, fourth-generation ALK inhibitors such as NVL-655 (Neladalkib) and TPX-0131 have been created. These agents were designed to exhibit “dual-mutation activity”, allowing them to simultaneously inhibit two co-occurring mutations within the ALK kinase domain[133]. TPX-0131 demonstrates strong central nervous system (CNS) penetration and binds tightly to the adenine region of the ATP-binding site of ALK kinase, thereby demonstrating strong activation. In preclinical studies, it showed potent inhibitory effects against a variety of ALK resistance mutations, particularly G1202R single mutations and L1196M/G1202R compound mutations[55]. TPX-0131 is over 100-fold more active against G1202R than Lorlatinib, though its activity against I1171N and G1269S is comparatively limited. In animal models, TPX-0131 achieved complete tumor regression and reached cerebrospinal fluid concentrations of up to 66% of plasma levels, confirming robust CNS penetration[139]. Despite these promising results, TPX-0131 was withdrawn from the Phase I trial (NCT04849273) due to an unsatisfactory risk-benefit profile. In contrast, NVL-655 is advancing more successfully in clinical development. It was designed to improve ALK selectivity while reducing off-target inhibition of the TRK family, which is expected to minimize CNS toxicity. NVL-655 exhibits potent inhibitory activity against multiple single and compound ALK resistance mutations, including G1202R, I1171N, G1202R/L1198F, and G1202R/L1196M, among others[140]. Importantly, its very low inhibitory activity against TrkB suggests a reduced risk of neurotoxicity. In mouse tumor models based on the EML4–ALK V1 fusion gene, NVL-655 demonstrated superior antitumor efficacy compared with Lorlatinib. The ongoing Phase I/II clinical trial ALKOVE-1 (NCT05384626) has reported favorable CNS penetration and manageable toxicity and side effects in heavily pretreated patients with ALK-positive NSCLC[141]. Phase I focused on dose escalation and safety, while Phase II is evaluating the objective response rate (ORR) as assessed by an independent review committee.
Beyond TPX-0131 and NVL-655, other novel ALK inhibitors, such as ZX-29, Gilteritinib, and dual-target compounds like CHMFL-ALK/EGFR-050 and CEP-37440, have shown promising preclinical efficacy. These agents are effective against diverse ALK resistance variants and may offer new therapeutic avenues, particularly for patients with compound mutations or brain metastases[142]. Nonetheless, these investigational drugs remain at an early stage of clinical development, and further studies are required to validate their efficacy and safety. Careful evaluation of potential side effects and long-term safety profiles will also be essential.
Combination therapy
Building on ALK-targeted drug therapy, additional treatment approaches such as chemotherapy, radiotherapy, and immunotherapy are increasingly being incorporated. With advances in understanding the mechanisms of lung cancer drug resistance and in molecular biology technologies, the treatment of NSCLC has become progressively more complex and diverse[143]. Developing treatment plans based on genetic information is essential for selecting the most appropriate strategies, both for ALK-TKIs and for combination regimens following the emergence of resistance[144]. Multidisciplinary collaboration and the accumulation of clinical expertise are also required to manage adverse events effectively[145]. In targeted therapy for lung cancer, it is essential to realize that tumor heterogeneity is dynamic, with ongoing changes in cancer-driving genes and epigenetic modifications. Therefore, repeated biopsies and comprehensive analyses of tumor cells and their microenvironment are particularly important to dynamically monitor the factors driving tumor progression.
CONCLUSION AND PROSPECT
Lung cancer is characterized by high morbidity and mortality and is prone to recurrence and metastasis, making early diagnosis and prognostic markers critically important. In epigenetic research, the identification of DNA hypermethylation patterns in transcription factor gene promoter sequences has revealed homeobox (HOX)-related genes that may serve as biomarkers for early detection and prognostic evaluation of lung cancer. In addition, dysregulated long non-coding RNAs (lncRNAs), such as HotTip, BLACAT1, and SOX2/ANRIL, have been detected in the serum of NSCLC patients. These circulating lncRNAs show potential as early diagnostic markers and may be associated with OS. Continued exploration of epigenetic mechanisms is likely to yield additional biomarkers, providing valuable guidance for improving early detection and prognostic assessment of NSCLC. Targeted therapy for ALK-positive NSCLC has significantly improved patient prognosis. However, the development of drug resistance has become a major clinical challenge. Current strategies to address resistance include performing re-biopsies to identify underlying mechanisms, adjusting treatment regimens, and investigating novel approaches such as combination targeted therapy and immunotherapy. Although second- and third-generation ALK-TKIs have achieved notable clinical success, resistance remains an unresolved problem.
In summary, resistance to ALK-targeted therapy in NSCLC is inevitable and multifactorial. Resistance mechanisms are highly heterogeneous and include ALK kinase domain mutations, bypass signaling activation, histological transformation, and immune escape. Recent studies have also highlighted the influence of the tumor immune microenvironment and checkpoint molecule regulation (e.g., PD-L1 upregulation) on resistance. Moreover, small cell transformation has emerged as a distinct and aggressive resistance phenotype that requires early detection and individualized management. Based on current knowledge, a comprehensive strategy that integrates dynamic monitoring of resistance mutations, molecular re-biopsy, and rational combination therapy (including next-generation ALK inhibitors, immunotherapy, or dual-target agents) is essential to improve long-term outcomes. Future research should aim to combine molecular profiling with immune landscape characterization to guide truly personalized treatment for ALK-positive NSCLC.
Overall, a deeper investigation into the molecular mechanisms of drug resistance and the immune microenvironment in ALK-positive NSCLC is crucial for designing more effective therapeutic strategies. Future efforts should focus not only on optimizing diagnosis and treatment at early stages but also on developing comprehensive treatment strategies that combine effective drugs with rational sequencing. As more next-generation drugs gain regulatory approval, their optimal clinical application must be carefully defined. At the same time, supportive care and toxicity management remain indispensable for maximizing treatment benefits and survival. Thus, future research should advance both fundamental understanding and clinical practice to achieve more precise and personalized therapies for patients with ALK-positive NSCLC.
DECLARATIONS
Acknowledgments
Figures were created with BioRender.com.
Authors’ contributions
Overall execution of the research and writing of the manuscript: Long M, Peng S
Data analysis: Wu Y
Provided critical insights into the research: Wen Q, Yin Z, Zhang X, Tan H, Xu Y
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by Major Research Programs for High-level Talents in Healthcare in Hunan Province (R2023184) and Hunan Provincial Administration of Traditional Chinese Medicine City State Joint Project(D2024017).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7-33.
2. Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247-53.
3. McCoach CE, Le AT, Gowan K, et al. Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non-small cell lung cancer. Clin Cancer Res. 2018;24:3334-47.
4. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175-80.
5. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561-6.
6. Wang S, Shi Y, Han X. [Advances in drug resistance mechanisms and prognostic markers of targeted therapy in ALK-positive non-small cell lung cancer]. Zhongguo Fei Ai Za Zhi. 2020;23:1014-22.
7. Shen J, Meng Y, Wang K, et al. EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell Signal. 2022;92:110264.
8. Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6:1118-33.
9. Solomon BJ, Liu G, Felip E, et al. Lorlatinib versus crizotinib in patients with advanced ALK-positive non-small cell lung cancer: 5-year outcomes from the Phase III CROWN study. J Clin Oncol. 2024;42:3400-9.
10. Lovly CM. New benchmark for targeted therapies in lung cancer: median progression-free survival for lorlatinib in advanced ALK+ non-small cell lung cancer surpasses 5 years. J Clin Oncol. 2024;42:3383-6.
11. Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374:54-61.
12. Toyokawa G, Inamasu E, Shimamatsu S, et al. Identification of a novel ALK G1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J Thorac Oncol. 2015;10:e55-7.
13. Michels SYF, Scheel AH, Wündisch T, et al.
14. Okada K, Araki M, Sakashita T, et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine. 2019;41:105-19.
15. Yanagitani N, Uchibori K, Koike S, et al. Drug resistance mechanisms in Japanese anaplastic lymphoma kinase-positive non-small cell lung cancer and the clinical responses based on the resistant mechanisms. Cancer Sci. 2020;111:932-9.
16. Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol. 2016;11:2027-32.
17. Ou SH, Greenbowe J, Khan ZU, et al. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer. 2015;88:231-4.
18. Ou SH, Milliken JC, Azada MC, Miller VA, Ali SM, Klempner SJ. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer. 2016;91:70-2.
19. Yoda S, Lin JJ, Lawrence MS, et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 2018;8:714-29.
20. Wang Y, He J, Xu M, et al. Holistic view of ALK TKI resistance in ALK-positive anaplastic large cell lymphoma. Front Oncol. 2022;12:815654.
21. Sharma GG, Mota I, Mologni L, Patrucco E, Gambacorti-Passerini C, Chiarle R. Tumor resistance against ALK targeted therapy-where it comes from and where it goes. Cancers. 2018;10:62.
22. Huang WS, Liu S, Zou D, et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem. 2016;59:4948-64.
23. Zhang S, Anjum R, Squillace R, et al. The potent ALK inhibitor brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res. 2016;22:5527-38.
24. Gainor JF, Chi AS, Logan J, et al. Alectinib dose escalation reinduces central nervous system responses in patients with anaplastic lymphoma kinase-positive non-small cell lung cancer relapsing on standard dose alectinib. J Thorac Oncol. 2016;11:256-60.
25. Redaelli S, Ceccon M, Zappa M, et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018;78:6866-80.
26. Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28:70-81.
27. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov. 2017;7:137-55.
28. Peters S, Zimmermann S. Management of resistance to first-line anaplastic lymphoma kinase tyrosine kinase inhibitor therapy. Curr Treat Options Oncol. 2018;19:37.
29. Cooper AJ, Sequist LV, Lin JJ. Author Correction: Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat Rev Clin Oncol. 2022;19:744.
30. Sampson J, Ju HM, Zhang N, Yeoh S, Choi J, Bayliss R. Targeting ERBB3 and AKT to overcome adaptive resistance in EML4-ALK-driven non-small cell lung cancer. Cell Death Dis. 2024;15:912.
31. Bauman JR, Liu G, Preeshagul I, et al. Real-world treatment sequencing and effectiveness of second- and third-generation ALK tyrosine kinase inhibitors for ALK-positive advanced non-small cell lung cancer. Lung Cancer. 2024;195:107919.
32. Liu L, Hou F, Liu Y, Li W, Zhang H. A case of lung adenocarcinoma response to alectinib harboring a rare EML4-ALK variant, Exon 6 of EML4 fused to Exon 18 of ALK. J Natl Compr Canc Netw. 2021;20:2-6.
33. Li X, Wang Z, Chen C, et al. A small-molecule degrader selectively inhibits the growth of ALK-rearranged lung cancer with ceritinib resistance. iScience. 2024;27:109015.
34. Gainor JF, Shaw AT. Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist. 2013;18:865-75.
35. Nam Y, Hwang D, Kim N, Seo HS, Selim KB, Sim T. Identification of 1H-pyrazolo[3,4-b]pyridine derivatives as potent ALK-L1196M inhibitors. J Enzyme Inhib Med Chem. 2019;34:1426-38.
36. Wang KL, Yeh TY, Hsu PC, et al. Discovery of novel anaplastic lymphoma kinase (ALK) and histone deacetylase (HDAC) dual inhibitors exhibiting antiproliferative activity against non-small cell lung cancer. J Enzyme Inhib Med Chem. 2024;39:2318645.
37. Gerlinger M, Norton L, Swanton C. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;369:1172-3.
38. Yang Y, Yang H, Gao Y, et al. EML4-ALK G1202R and EML4-ALK L1196M mutations induce crizotinib resistance in non-small cell lung cancer cells through activating epithelial-mesenchymal transition mediated by MDM2/MEK/ERK signal axis. Cell Biol Int. 2025;49:55-67.
39. Leporati R, Miliziano D, Beninato T, et al. Response to lorlatinib rechallenge in a case of ALK-rearranged metastatic NSCLC with a resistance mutation to second generation TKIs. Tumori. 2024;110:NP1-4.
40. Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662-73.
41. Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17.
42. Li Y, Hao Z, Ma Y, et al. Alectinib continuation beyond progression in ALK-positive non-small cell lung cancer with alectinib-refractory. Transl Lung Cancer Res. 2024;13:152-62.
43. Han R, Lu CH, Hu C, et al. Brigatinib, a newly discovered AXL inhibitor, suppresses AXL-mediated acquired resistance to osimertinib in EGFR-mutated non-small cell lung cancer. Acta Pharmacol Sin. 2024;45:1264-75.
44. Chapman AM, Sun KY, Ruestow P, Cowan DM, Madl AK. Lung cancer mutation profile of EGFR, ALK, and KRAS: meta-analysis and comparison of never and ever smokers. Lung Cancer. 2016;102:122-34.
45. Yamada T, Takeuchi S, Nakade J, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res. 2012;18:3592-602.
46. Ruiz CF, Montal ED, Haley JA, Bott AJ, Haley JD. SREBP1 regulates mitochondrial metabolism in oncogenic KRAS expressing NSCLC. FASEB J. 2020;34:10574-89.
47. Ma X, Ma Z, Qi X, et al. Identification of a novel Src inhibitor K882 derived from quinazoline-based stilbenes with anti-NSCLC effect. Bioorg Chem. 2025;156:108185.
48. Janostiak R, Malvi P, Wajapeyee N. Anaplastic lymphoma kinase confers resistance to BRAF kinase inhibitors in melanoma. iScience. 2019;16:453-67.
49. Dong X, Fernandez-Salas E, Li E, Wang S. Elucidation of resistance mechanisms to second-generation ALK inhibitors alectinib and ceritinib in non-small cell lung cancer cells. Neoplasia. 2016;18:162-71.
50. Urbanska EM, Sørensen JB, Melchior LC, Costa JC, Santoni-Rugiu E. Changing ALK-TKI-resistance mechanisms in rebiopsies of ALK-rearranged NSCLC: ALK- and BRAF-mutations followed by epithelial-mesenchymal transition. Int J Mol Sci. 2020;21:2847.
51. Zhuang X, Zhao C, Li J, et al. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med. 2019;8:2858-66.
52. Talwelkar SS, Mäyränpää MI, Schüler J, et al. PI3Kβ inhibition enhances ALK-inhibitor sensitivity in ALK-rearranged lung cancer. Mol Oncol. 2023;17:747-64.
53. Hao XL, Gao LY, Deng XJ, et al. Identification of TC2N as a novel promising suppressor of PI3K-AKT signaling in breast cancer. Cell Death Dis. 2019;10:424.
54. Smiles WJ, Catalano L, Stefan VE, Weber DD, Kofler B. Metabolic protein kinase signalling in neuroblastoma. Mol Metab. 2023;75:101771.
55. Xie J, Gao Y, Xu W, Zhu J. Mechanisms of resistance to ALK inhibitors and corresponding treatment strategies in lung cancer. Int J Gen Med. 2025;18:2151-71.
56. Li Y, Li Y, Zhang H, et al. EML4-ALK-mediated activation of the JAK2-STAT pathway is critical for non-small cell lung cancer transformation. BMC Pulm Med. 2021;21:190.
57. Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121-34.
58. Conlon TM, John-Schuster G, Heide D, et al. Inhibition of LTβR signalling activates WNT-induced regeneration in lung. Nature. 2021;588:151-6.
59. Lin Y, Higashisaka K, Shintani T, et al. Progesterone receptor membrane component 1 leads to erlotinib resistance, initiating crosstalk of Wnt/β-catenin and NF-κB pathways, in lung adenocarcinoma cells. Sci Rep. 2020;10:4748.
60. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2020;68:394-424.
61. Clamon G, Zeitler W, An J, Hejleh TA. Transformational changes between non-small cell and small cell lung cancer-biological and clinical relevance - a review. Am J Clin Oncol. 2020;43:670-5.
62. Yin X, Li Y, Wang H, et al. Small cell lung cancer transformation: from pathogenesis to treatment. Semin Cancer Biol. 2022;86:595-606.
63. Socinski MA, Morris DE, Halle JS, et al. Induction and concurrent chemotherapy with high-dose thoracic conformal radiation therapy in unresectable stage IIIA and IIIB non-small-cell lung cancer: a dose-escalation phase I trial. J Clin Oncol. 2004;22:4341-50.
64. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16:e165-72.
65. Fujita S, Masago K, Katakami N, Yatabe Y. Transformation to SCLC after treatment with the ALK inhibitor alectinib. J Thorac Oncol. 2016;11:e67-72.
66. Miyamoto S, Ikushima S, Ono R, et al. Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib. Jpn J Clin Oncol. 2016;46:170-3.
67. Roca E, Gurizzan C, Amoroso V, Vermi W, Ferrari V, Berruti A. Outcome of patients with lung adenocarcinoma with transformation to small-cell lung cancer following tyrosine kinase inhibitors treatment: a systematic review and pooled analysis. Cancer Treat Rev. 2017;59:117-22.
68. Marcoux N, Gettinger SN, O’Kane G, et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J Clin Oncol. 2019;37:278-85.
69. Imakita T, Fujita K, Kanai O, et al. Small cell transformation of non-small cell lung cancer under immunotherapy: case series and literature review. Thorac Cancer. 2021;12:3062-7.
70. Levacq D, D’Haene N, de Wind R, Remmelink M, Berghmans T. Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: a new mechanism of resistance to ALK inhibitors. Lung Cancer. 2016;102:38-41.
71. Hobeika C, Rached G, Eid R, et al. ALK-rearranged adenocarcinoma transformed to small-cell lung cancer: a new entity with specific prognosis and treatment? Per Med. 2018;15:111-5.
72. Zhou N, Leung CH, William WN Jr, et al. Impact of select actionable genomic alterations on efficacy of neoadjuvant immunotherapy in resectable non-small cell lung cancer. J Immunother Cancer. 2024;12:e009677.
73. Zhang D, Wang M, Liu G, et al. Novel FABP4+C1q+ macrophages enhance antitumor immunity and associated with response to neoadjuvant pembrolizumab and chemotherapy in NSCLC via AMPK/JAK/STAT axis. Cell Death Dis. 2024;15:717.
74. Lu C, Gao Z, Wu D, et al. Understanding the dynamics of TKI-induced changes in the tumor immune microenvironment for improved therapeutic effect. J Immunother Cancer. 2024;12:e009165.
75. Zhang B, Zeng J, Zhang H, et al. Characteristics of the immune microenvironment and their clinical significance in non-small cell lung cancer patients with ALK-rearranged mutation. Front Immunol. 2022;13:974581.
76. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585-93.
77. Budczies J, Kirchner M, Kluck K, et al. Deciphering the immunosuppressive tumor microenvironment in ALK- and EGFR-positive lung adenocarcinoma. Cancer Immunol Immunother. 2022;71:251-65.
78. Liu SY, Dong ZY, Wu SP, et al. Clinical relevance of PD-L1 expression and CD8+ T cells infiltration in patients with EGFR-mutated and ALK-rearranged lung cancer. Lung Cancer. 2018;125:86-92.
79. Horvat NK, Chocarro S, Marques O, et al. Superparamagnetic iron oxide nanoparticles reprogram the tumor microenvironment and reduce lung cancer regrowth after crizotinib treatment. ACS Nano. 2024;18:11025-41.
80. Camidge DR, Doebele RC, Kerr KM. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat Rev Clin Oncol. 2019;16:341-55.
81. Gu D, Hu L, Huang S, Guo L. Expression and clinical significance of programmed death ligand-1 evaluated by 22C3 antibody in pleural effusion metastatic non-small-cell lung cancer. Cytojournal. 2024;21:70.
82. Peng S, Wang R, Zhang X, et al. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. 2019;18:165.
83. Qi R, Yu Y, Shen M, Lv D, He S. Current status and challenges of immunotherapy in ALK rearranged NSCLC. Front Oncol. 2022;12:1016869.
84. Zhang Q, Zhang Y, Chen Y, Qian J, Zhang X, Yu K. A novel mTORC1/2 inhibitor (MTI-31) inhibits tumor growth, epithelial-mesenchymal transition, metastases, and improves antitumor immunity in preclinical models of lung cancer. Clin Cancer Res. 2019;25:3630-42.
85. Spigel DR, Reynolds C, Waterhouse D, et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation - positive advanced non-small cell lung cancer (CheckMate 370). J Thorac Oncol. 2018;13:682-8.
86. Golding B, Luu A, Jones R, Viloria-Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol Cancer. 2018;17:52.
87. Furugaki K, Fujimura T, Sakaguchi N, et al. Combined blockade of GPX4 and activated EGFR/HER3 bypass pathways inhibits the development of ALK-inhibitor-induced tolerant persister cells in ALK-fusion-positive lung cancer. Mol Oncol. 2025;19:519-39.
88. Hong S, Chen N, Fang W, et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology. 2016;5:e1094598.
89. Ota K, Azuma K, Kawahara A, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21:4014-21.
90. Du P, Hu T, An Z, Li P, Liu L.
91. Felip E, de Braud FG, Maur M, et al. Ceritinib plus nivolumab in patients with advanced ALK-rearranged non-small cell lung cancer: results of an open-label, multicenter, phase 1B study. J Thorac Oncol. 2020;15:392-403.
92. Gan GN, Weickhardt AJ, Scheier B, et al. Stereotactic radiation therapy can safely and durably control sites of extra-central nervous system oligoprogressive disease in anaplastic lymphoma kinase-positive lung cancer patients receiving crizotinib. Int J Radiat Oncol Biol Phys. 2014;88:892-8.
93. Chen Y, Ma G, Su C, et al. Apatinib reverses alectinib resistance by targeting vascular endothelial growth factor receptor 2 and attenuating the oncogenic signaling pathway in echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase fusion gene-positive lung cancer cell lines. Anticancer Drugs. 2018;29:935-43.
94. Choudhury NJ, Young RJ, Sellitti M, Miller A, Drilon A. Lorlatinib and bevacizumab activity in ALK-rearranged lung cancers after lorlatinib progression. JCO Precis Oncol. 2020;4:PO.20.00271.
95. Choi SH, Kim DH, Choi YJ, et al. Multiple receptor tyrosine kinase activation related to ALK inhibitor resistance in lung cancer cells with ALK rearrangement. Oncotarget. 2017;8:58771-80.
96. Miyawaki M, Yasuda H, Tani T, et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol Cancer Res. 2017;15:106-14.
97. Tani T, Yasuda H, Hamamoto J, et al. Activation of EGFR bypass signaling by TGFα overexpression induces acquired resistance to alectinib in ALK-translocated lung cancer cells. Mol Cancer Ther. 2016;15:162-71.
98. Kang J, Chen HJ, Zhang XC, et al. Heterogeneous responses and resistant mechanisms to crizotinib in ALK-positive advanced non-small cell lung cancer. Thorac Cancer. 2018;9:1093-103.
99. Hrustanovic G, Olivas V, Pazarentzos E, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015;21:1038-47.
101. Shrestha N, Bland AR, Bower RL, Rosengren RJ, Ashton JC. Inhibition of mitogen-activated protein kinase kinase alone and in combination with anaplastic lymphoma kinase (ALK) inhibition suppresses tumor growth in a mouse model of ALK-positive lung cancer. J Pharmacol Exp Ther. 2020;374:134-40.
102. Shrestha N, Nimick M, Dass P, Rosengren RJ, Ashton JC. Mechanisms of suppression of cell growth by dual inhibition of ALK and MEK in ALK-positive non-small cell lung cancer. Sci Rep. 2019;9:18842.
103. Tanizaki J, Okamoto I, Takezawa K, et al. Combined effect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell lung cancer cells. Br J Cancer. 2012;106:763-7.
104. Jänne PA, Shaw AT, Camidge DR, et al. Combined Pan-HER and ALK/ROS1/MET inhibition with dacomitinib and crizotinib in advanced non-small cell lung cancer: results of a phase I study. J Thorac Oncol. 2016;11:737-47.
105. Liu QG, Wu J, Wang ZY, et al. ALK-based dual inhibitors: focus on recent development for non-small cell lung cancer therapy. Eur J Med Chem. 2025;291:117646.
106. Jiang C, Xie S, Jia K, Feng G, Ren X, Wang Y. Exploring cellular plasticity and resistance mechanisms in lung cancer: Innovations and emerging therapies. J Pharm Anal. 2025;15:101179.
107. Yamaguchi N, Lucena-Araujo AR, Nakayama S, et al. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer. 2014;83:37-43.
108. Rossing HH, Grauslund M, Urbanska EM, et al. Concomitant occurrence of EGFR (epidermal growth factor receptor) and KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutations in an ALK (anaplastic lymphoma kinase)-positive lung adenocarcinoma patient with acquired resistance to crizotinib: a case report. BMC Res Notes. 2013;6:489.
109. Sahnane N, Frattini M, Bernasconi B, et al. EGFR and KRAS mutations in ALK-positive lung adenocarcinomas: biological and clinical effect. Clin Lung Cancer. 2016;17:56-61.
110. Sweis RF, Thomas S, Bank B, Fishkin P, Mooney C, Salgia R. Concurrent EGFR mutation and ALK translocation in non-small cell lung cancer. Cureus. 2016;8:e513.
111. Yang JJ, Zhang XC, Su J, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res. 2014;20:1383-92.
112. Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. 2018;15:150.
113. Lehman A, Perissinotti A, Aitken S. Cytomegalovirus viremia and hepatitis B reactivation in patient with RET fusion-positive non-small cell lung cancer treated with pralsetinib. J Oncol Pharm Pract. 2025;31:674-8.
114. Zheng Q, Fang W, Huang Y, Gan J, Zhang L. Identification of a novel KIF5B-RET, ABHD17C-RET double-fusion variant in lung adenocarcinoma and response to cabozantinib. J Thorac Oncol. 2020;15:e132-3.
115. Song Z, Xia Z, Ji Y, et al. An orally available tyrosine kinase ALK and RET dual inhibitor bearing the tetracyclic benzo[b]carbazolone core. Eur J Med Chem. 2016;118:244-9.
116. Chen JF, Guo SJ, He B, et al. Advances of dual inhibitors based on ALK for the treatment of cancer. Bioorg Chem. 2025;159:108417.
117. Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375-7.
118. Parvaresh H, Roozitalab G, Golandam F, Behzadi P, Jabbarzadeh Kaboli P. Unraveling the potential of ALK-targeted therapies in non-small cell lung cancer: comprehensive insights and future directions. Biomedicines. 2024;12:297.
119. Li MSC, Mok KKS, Mok TSK. Developments in targeted therapy & immunotherapy-how non-small cell lung cancer management will change in the next decade: a narrative review. Ann Transl Med. 2023;11:358.
120. Hu Z, Wang N, Zhang Y, et al. PD-L1 mRNA derived from tumor-educated platelets as a potential immunotherapy biomarker in non-small cell lung cancer. Transl Lung Cancer Res. 2024;13:345-54.
121. Cavazzoni A, Digiacomo G, Volta F, et al. PD-L1 overexpression induces STAT signaling and promotes the secretion of pro-angiogenic cytokines in non-small cell lung cancer (NSCLC). Lung Cancer. 2024;187:107438.
122. Reck M, Mok TSK, Nishio M, et al; IMpower150 Study Group. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7:387-401.
123. Gaissmaier L, Christopoulos P. Immune modulation in lung cancer: current concepts and future strategies. Respiration. 2021;99:903-29.
124. Wang Q, Su C, Zhou C. Recent advances in immunotherapy for lung cancer. Cancer Innov. 2023;2:18-24.
125. Furugaki K, Fujimura T, Mizuta H, et al. FGFR blockade inhibits targeted therapy-tolerant persister in basal FGFR1- and FGF2-high cancers with driver oncogenes. NPJ Precis Oncol. 2023;7:107.
126. Haderk F, Chou YT, Cech L, et al. Focal adhesion kinase-YAP signaling axis drives drug-tolerant persister cells and residual disease in lung cancer. Nat Commun. 2024;15:3741.
127. Huang S, Li C, Armstrong EA, et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73:824-33.
128. Hao Y, Li B, Huang D, et al. Developing a semi-supervised approach using a PU-learning-based data augmentation strategy for multitarget drug discovery. Int J Mol Sci. 2024;25:8239.
129. Xu L, Kikuchi E, Xu C, et al. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res. 2012;72:3302-11.
130. Iacono D, Chiari R, Metro G, et al. Future options for ALK-positive non-small cell lung cancer. Lung Cancer. 2015;87:211-9.
131. Ramalingam SS, Vansteenkiste J, Planchard D, et al; FLAURA Investigators. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41-50.
132. Jeon H, Wang S, Song J, Gill H, Cheng H. Update 2025: management of non‑small-cell lung cancer. Lung. 2025;203:53.
133. Voena C, Ambrogio C, Iannelli F, Chiarle R. ALK in cancer: from function to therapeutic targeting. Nat Rev Cancer. 2025;25:359-78.
135. Yang Y, Min J, Yang N, et al. Envonalkib versus crizotinib for treatment-naive ALK-positive non-small cell lung cancer: a randomized, multicenter, open-label, phase III trial. Signal Transduct Target Ther. 2023;8:301.
136. Recondo G, Mezquita L, Facchinetti F, et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin Cancer Res. 2020;26:242-55.
137. Wang J, Liu B, Zheng Q, Xiao R, Chen J. Newly emerged ROS1 rearrangement in a patient with lung adenocarcinoma following resistance to immune checkpoint inhibitors: a case report. Front Oncol. 2024;14:1507658.
138. Liao Y, Remsing Rix LL, Li X, et al. Differential network analysis of ROS1 inhibitors reveals lorlatinib polypharmacology through co-targeting PYK2. Cell Chem Biol. 2024;31:284-97.e10.
139. Murray BW, Zhai D, Deng W, et al. TPX-0131, a potent CNS-penetrant, next-generation inhibitor of wild-type ALK and ALK-resistant mutations. Mol Cancer Ther. 2021;20:1499-507.
140. Ou SI, Nagasaka M, Brazel D, Hou Y, Zhu VW. Will the clinical development of 4th-generation “double mutant active” ALK TKIs (TPX-0131 and NVL-655) change the future treatment paradigm of ALK+ NSCLC? Transl Oncol. 2021;14:101191.
141. Lin JJ, Horan JC, Tangpeerachaikul A, et al. NVL-655 is a selective and brain-penetrant inhibitor of diverse ALK-mutant oncoproteins, including lorlatinib-resistant compound mutations. Cancer Discov. 2024;14:2367-86.
142. Pulte ED, Norsworthy KJ, Wang Y, et al. FDA Approval Summary: gilteritinib for relapsed or refractory acute myeloid leukemia with a FLT3 mutation. Clin Cancer Res. 2021;27:3515-21.
143. Nishio M, Kim DW, Wu YL, et al. Crizotinib versus chemotherapy in Asian patients with ALK-positive advanced non-small cell lung cancer. Cancer Res Treat. 2018;50:691-700.
144. Yang Q, Sun K, Gao T, et al. SIRT1 silencing promotes EMT and crizotinib resistance by regulating autophagy through AMPK/mTOR/S6K signaling pathway in EML4-ALK L1196M and EML4-ALK G1202R mutant non-small cell lung cancer cells. Mol Carcinog. 2024;63:2133-44.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.