


Decoding donor/acceptor hierarchy in DAD triads via fragment-centric machine learning
 0
0 
Abstract
The rational design of donor–acceptor–donor (DAD) triads is often hindered by the complex, nonlinear interactions between molecular fragments. This study develops an interpretable machine learning (ML) framework utilizing fragment orbital descriptors (FODs) to predict key energetic criteria of hot-exciton materials and establish quantitative structure-property relationships. Through a synergy of micro-interpretability and macro-statistical analysis, we uncover a definitive fragment-selectivity hierarchy: acceptor molecular orbitals act as the primary regulators, contributing over 70% to energy gap determination, while donor fragments serve as secondary modulators for fine-tuning. We identify specific chemical manifestations of this hierarchy: DPP-based acceptors preferentially maximize the triplet-triplet energy gap (ΔETT) through enhanced high-lying triplet separation, whereas MI-containing systems minimize the singlet-triplet energy gap (ΔEST) via optimized singlet-triplet coupling. This work provides a deterministic informatics framework that decodes the “fragment-to-whole” regulation logic, offering practical guidelines for the high-throughput discovery and precision engineering of high-performance optoelectronic materials.
Keywords
INTRODUCTION
Rapid advancements in artificial intelligence (AI) and the availability of massive datasets have ushered in a transformative paradigm for scientific discovery through interdisciplinary integration[1-5]. This trend has profoundly impacted materials science, where data-driven methodologies grounded in the “Big Data + AI” framework have significantly accelerated the exploration of complex chemical spaces[6-8]. In particular, machine learning (ML)[9], a core AI technology, has emerged as a powerful computational tool for predicting material properties and uncovering fundamental structure-property relationships across diverse systems such as perovskites[10,11], alloys[12,13], and organic light-emitting diodes (OLEDs)[14-17]. ML approaches not only enhance the efficiency of precise materials design but also provide unique insights into hidden patterns within complex material datasets[1].
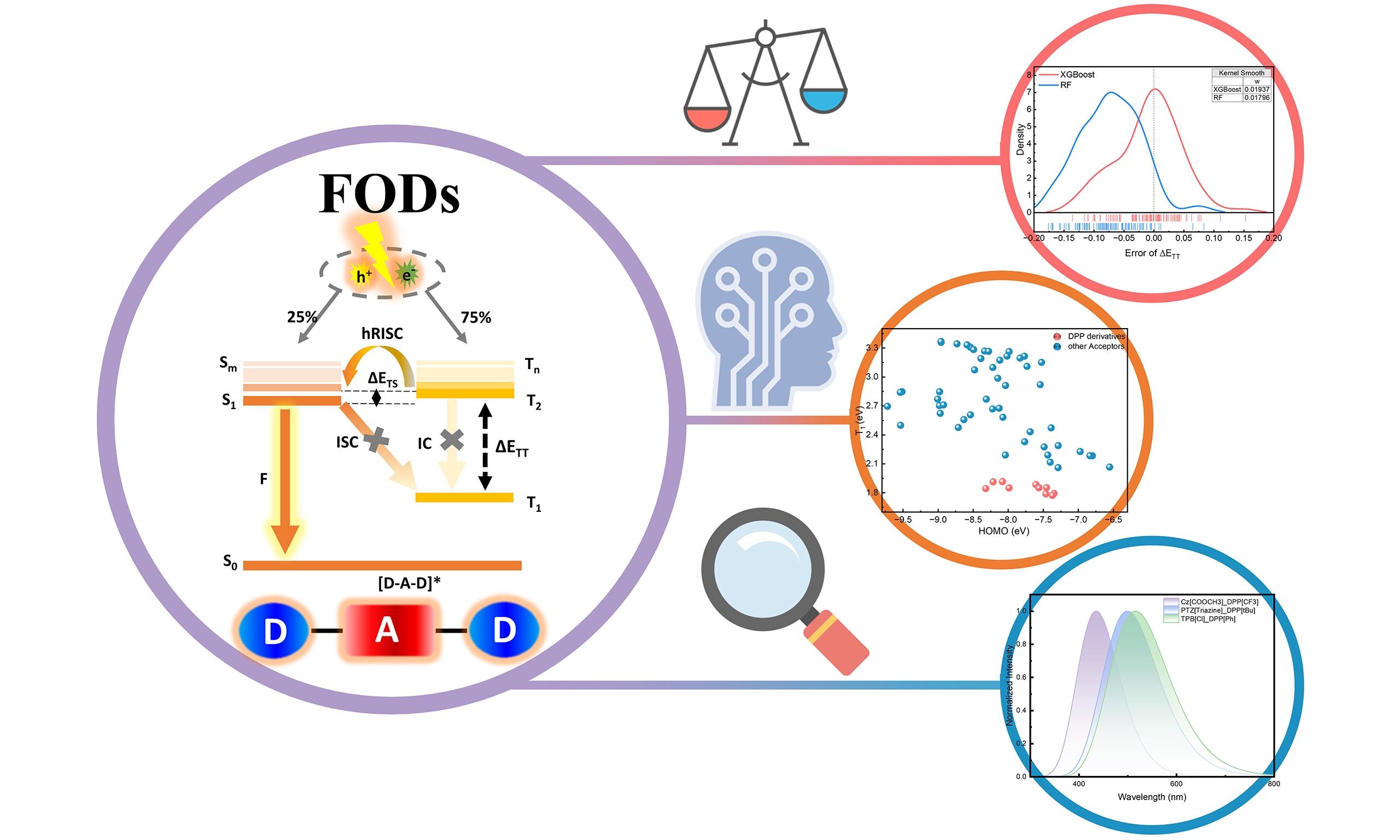
Among various classes of OLED materials, hot-exciton compounds have attracted considerable attention due to their distinctive photophysical properties, specifically their ability to harvest triplet excitons through high-lying reverse intersystem crossing (hRISC)[18-21]. A characteristic feature of these materials is the hybridized local and charge-transfer (HLCT) excited state, significantly enhancing oscillator strength and luminescence efficiency[22]. Among these, donor–acceptor–donor (D–A–D, abbreviated as DAD) triads have demonstrated particularly advantageous exciton management capabilities, enabled by minimized energy gaps (ET2 - ES1, ΔETS) between high-lying triplet (Tn) and singlet (Sm) states, and by stabilized HLCT states originating from the synergistic electronic interactions between donor and acceptor fragments, typically as hot exciton material is constructed from donor (D) and acceptor (A) molecular fragments that self-assemble into characteristic D–A, D–A–D, or D–A–D′ structures[18,23]. The intrinsic strength of the DAD molecular architecture lies in the strong through-bond electronic coupling mediated by central acceptor fragments, which concurrently enhances device efficiency, exciton utilization efficiency (EUE), and operational stability[24]. Crucially, rapid hRISC process enables simultaneous harvesting of singlet and triplet excitons while mitigating long-term degradation through suppressed exciton quenching[25]. Beyond the intrinsically small ΔETS, these advantages of DADs can be also governed by other two essential energy criteria as per Kasha’s rule [Figure 1][26]: (i) small ΔEST (ES1 - ET1) between emissive Sm (typically S1) and Tn (typically T1) states, which accelerates the RISC process and further boosts the EUE[27,28]; (ii) large ΔETT (ET2 - ET1) between higher-lying Tn (n ≥ 2) and T1 states, inhibiting triplet-triplet annihilation via internal conversion and thus preventing efficiency roll-off[29,30]. These energy gap requirements establish fundamental guidelines for the rational design and selection of DAD molecular architectures[26].
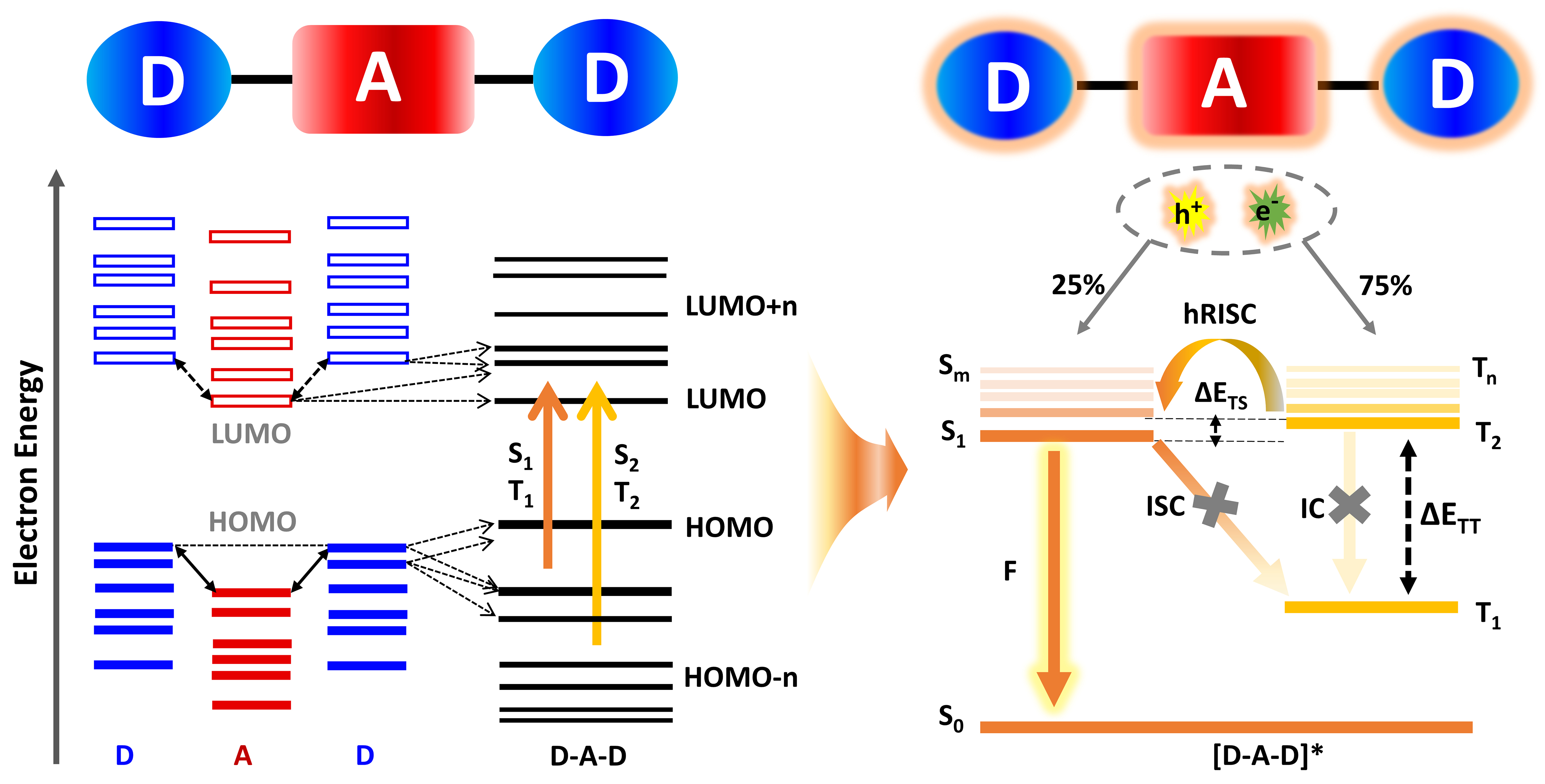
Figure 1. The molecular orbitals distributions of both the fragments (donor and acceptor) and the triads (DAD) under the ground states and excited states by electroluminescence of “hot-exciton” TADF materials. DAD: Donor–acceptor–donor; TADF: thermally activated delayed fluorescence; LUMO: lowest occupied molecular orbital; HOMO: highest occupied molecular orbital; hRISC: high-lying reverse intersystem crossing; IC: internal conversion.
Despite their promising attributes, rationally designing DAD molecules remains challenging due to their complex molecular architectures and demanding synthetic processes[18]. Identifying optimal donor/acceptor (D/A) combinations that satisfy the critical energy gap criteria (ΔETT, ΔEST) through conventional trial-and-error approaches is both time-consuming and resource-intensive. While density-functional theory (DFT) calculations can theoretically predict these key parameters, the molecular complexity of DADs still makes such methods computationally prohibitive including expensive fees for high-throughput screening and exhaustive analysis. These barriers of synthetic and computational complexity highlights the need for more efficient approaches.
In this context, ML offers an ideal solution, enabling rapid prediction of critical photophysical properties such as ΔETT and ΔEST[1,31-33], thereby facilitating efficient screening of optimal D/A combinations and elucidating underlying structure-property relationships[1,34,35]. While prior studies have successfully applied ML to OLED materials, systematic ML exploration specifically targeting the unique energy-gap characteristics of DADs remains limited such as fused tricyclic compounds[36], phosphorescent molecules[37], and emitting layer host materials[16]. In our previous work, a route to accelerate the discovery of DAD candidates by virtue of D/A fragments has been provided with evaluation of energy gaps through TDDFT/TDA computations[25]. Although the results identified a series of promising candidates with promoting hRISC and minimizing competing triplet-triplet IC, the ab-initio computations based on quantum mechanics (QM) and chemistry programs are much more expensive than ML acceleration.
Here, we propose an innovative ML-driven approach utilizing fragment orbital descriptors (FODs) to predict key excited-state energy gaps (ΔETT, ΔEST, and ΔETS) in DAD triads via a Fragment-to-Whole molecular prediction strategy with a per-molecule CPU cost on the order of seconds. Leveraging a dataset of 5,400 DAD molecules derived from quantum chemical calculations, we establish accurate predictive models and unravel detailed fragment-property correlations through comprehensive interpretable ML analyses. This dual strategy not only accelerates the identification of high-performance OLED candidates but also provides fundamental insights into how fragment-level properties control macroscopic energy gap characteristics, thereby offering concrete theoretical guidelines for rational DAD design.
MATERIALS AND METHODS
As shown in Figure 1, enhancing EUE requires optimizing two key energy gaps for DADs: small ΔEST and large ΔETT. To achieve efficient and accurate prediction of these parameters by virtue of ML, we choose the descriptors for ML based on frontier molecular orbital (FMO) theory, which governs molecular properties through interactions between the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals, thereby determining electron distributions and energy characteristics. Given the proven effectiveness of FMO-based descriptors in prior studies[36-40] and recognizing that the photophysical behavior of DADs arise from D/A fragment interactions, we employ the “Fragment-To-Whole” strategy[41], using the HOMO, LUMO, HOMO-1, and LUMO+1 of individual D/A fragments as key descriptors to predict whole-molecule energy gap efficiently and interpretably, marked as HD, LD, H1D, L1D, HA, LA, H1A, L1A and called as FODs.
While exploring the connections between D/A fragment orbitals and DAD molecular energy characteristics is crucial, we must first validate that our chosen descriptors can effectively predict these energy gaps through ML. This necessitates constructing a robust database - a challenging task given the limited availability of experimental and theoretical DAD data. To address this, we adopted a usual computational approach where simpler properties are calculated by traditional ways to predict more complex ones via ML[42]. Building upon our previous work[25], we systematically assembled a comprehensive DAD library by combining: six high-probability acceptors (PT, MI, DPP, Ant, BZP, BZ), nine promising donor derivatives (TPA, DPA, PhCz, Pyr, PTZ, TPB, DMF, NPA, Cz), and ten electronically diverse substituents (The structures of the derivatives and the substituent groups are shown in Figure 2), ultimately generating 5,400 unique DAD structures for prediction and analysis. Besides, an extended training set of 1,011 DAD molecules with varied D/A configurations from the prior study[25] is also established for ML training. All molecular entries include both FOD and corresponding DADs’ energy gaps calculated at the wB97X-D/6-31G(d)[43] level by DFT method, which is chosen for its demonstrated accuracy in modeling DAD systems. The meticulous selection of basis sets and functionals is critical to ensuring the fidelity of FODs and energy gaps, as these parameters directly dictate the quality and predictive accuracy of the resulting ML models.
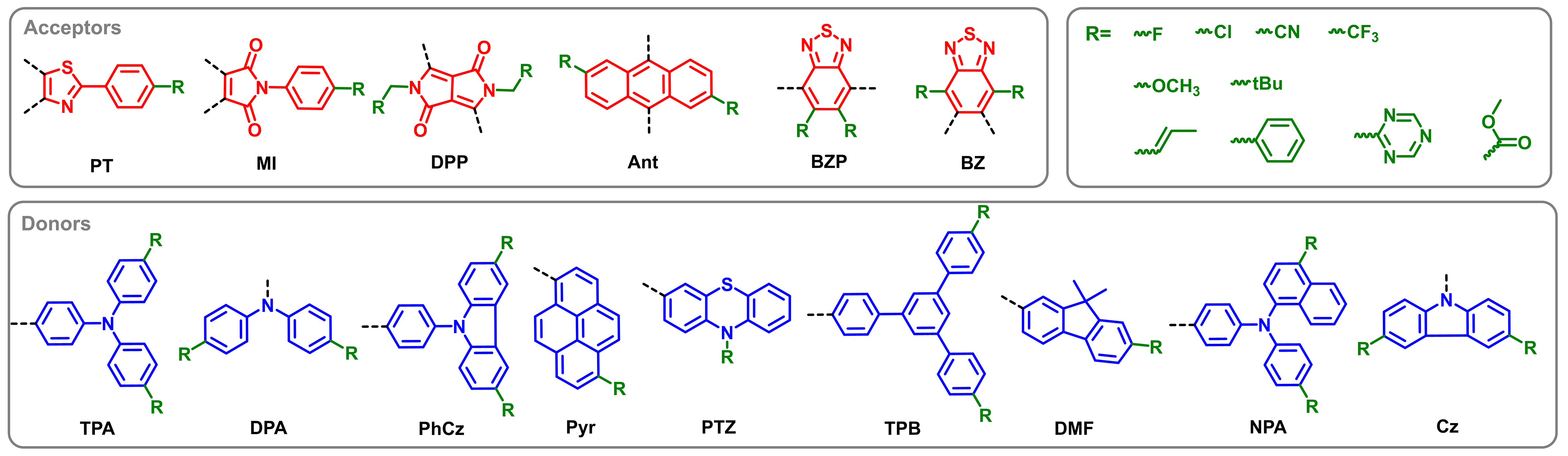
Figure 2. Chemical structures of acceptors (in red) and donors (in blue) derivatives with the substituent groups (in green) studied in this work.
With the database established, we proceed to construct ML models to predict ΔETT/ΔEST and to capture the nonlinear structure-property relationships. Given the moderate size of our dataset, for each target we implement eight distinct ML algorithms encompassing diverse theoretical frameworks: random forest (RF), eXtreme gradient boosting (XGBoost), linear regression (LR), ridge regression, K-nearest neighbors (KNN) regression, decision tree (DT) regression, support vector regression (SVR), and multilayer perceptron (MLP), all executed using Python’s scikit-learn package. The dataset is partitioned into training and testing sets with an 8:2 ratio, and model performance is evaluated using both root mean square error (RMSE) and the coefficient of determination (R2). To optimize each model while maintaining computational efficiency, we employed a hybrid hyperparameter tuning strategy combining random search with grid search, complemented by five-fold cross-validation during the training process to enhance model robustness and prevent overfitting.
Based on comparative performance, both RF and XGBoost emerge as superior predictors and are subsequently employed to predict ΔETT and ΔEST across all 5,400 DAD molecules. To leverage their complementary strengths - RF utilizing bagging method and XGBoost employing boosting method - we adopt an ensemble approach by averaging their predictions. This strategy effectively minimizes potential biases inherent to either individual model while capitalizing on their collective predictive power for more reliable results.
To elucidate the fundamental relationships between DADs’ energy gaps and D/A fragment orbitals (FODs), we employed interpretability approaches of ML. At the microscopic level, we conduct partial dependence plots (PDP), individual conditional expectation (ICE) analyses, and SHapley Additive exPlanations (SHAP) to reveal the quantum mechanical interactions governing these structure-property relationships. In parallel, macroscopic statistical analyses are performed to establish general trends in fragment contributions. These multiscale investigations can reveal distinct fragment selectivity patterns, demonstrating that specific D/A fragments consistently produce characteristic energy-level characteristics in the resulting DADs. The derived design principles not only provide physical insights into molecular formation mechanisms but also establish practical guidelines for the rational design of DAD systems with targeted optoelectronic properties such as energy gaps.
RESULTS AND DISCUSSION
ML results of FODs
Figure 3 presents the ML prediction results using FODs for two target attributes (ΔETT and ΔEST). Figure 3A and B demonstrate that both RF and XGBoost significantly outperform other models, achieving high R2 values (> 0.90) and low RMSE (< 0.1 eV), with DT showing competitive but slightly inferior performance. This superior predictive capability stems from their ensemble nature - while employing DT as base learners, RF (using bagging) and XGBoost (using boosting) integrate multiple weak predictors to enhance overall performance.
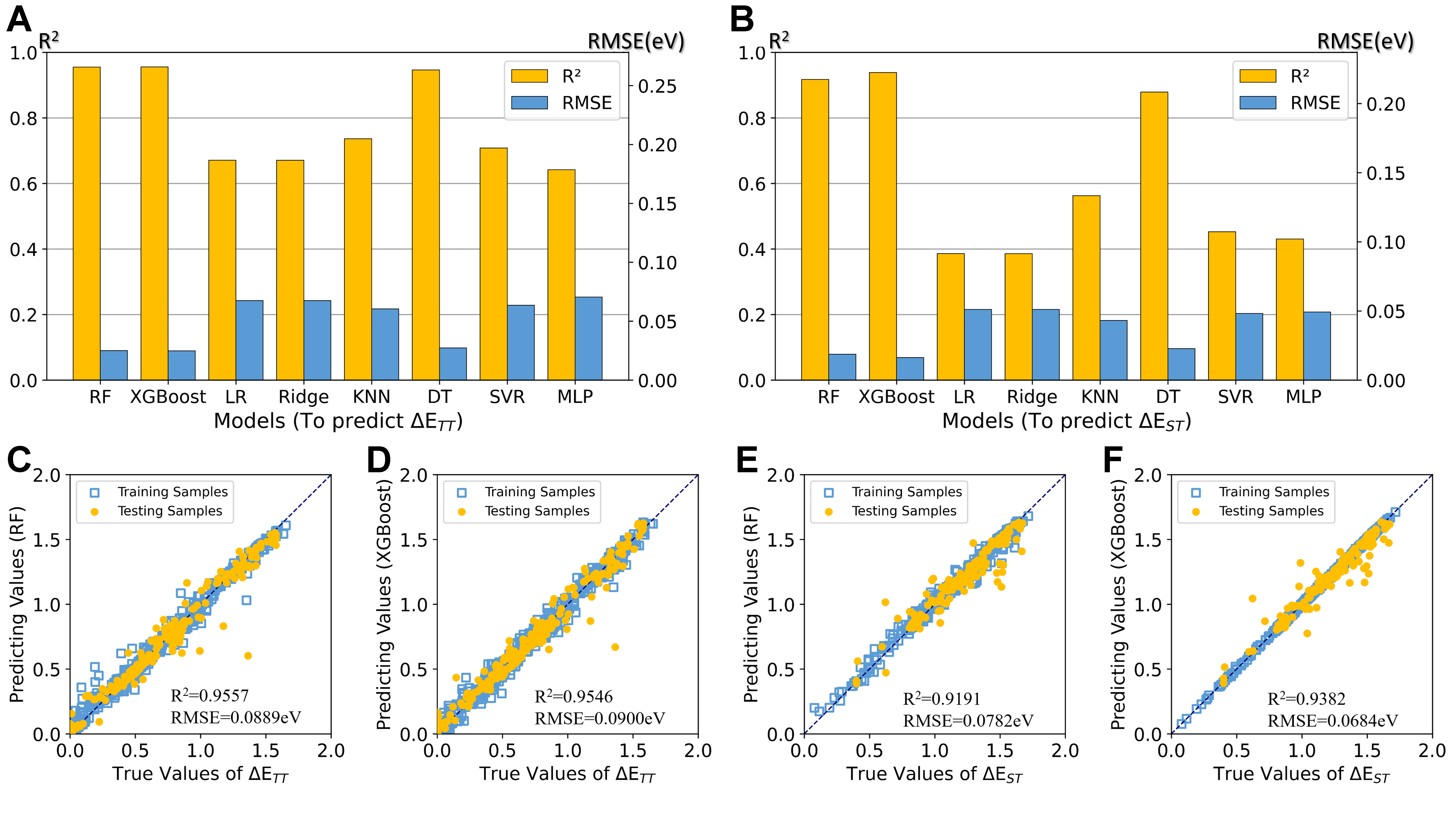
Figure 3. ML performance using FODs for predicting target attributes ΔETT and ΔEST. (A and B) Performance metrics (R2 in orange, RMSE in blue) across eight ML algorithms; (C-F) Predicted vs. actual values for RF and XGBoost models on both training and testing samples, demonstrating the prediction accuracy for attributes ΔETT (C and D) and ΔEST (E and F). The diagonal dashed lines represent ideal 1:1 correspondence. ML: Machine learning; FODs: fragment orbital descriptors; R2: the coefficient of determination; RMSE: root mean square error; RF: random forest; XGBoost: eXtreme gradient boosting; LR: linear regression; KNN: K-nearest neighbors; DT: decision tree; SVR: support vector regression; MLP: multilayer perceptron.
The model validation results in Figure 3C-F reveal excellent agreement between predicted and actual values for both training and testing sets, with no apparent overfitting. Although predictions for attribute ΔETT show marginally better accuracy than attribute ΔEST, the consistently high R2 (> 90%) across testing sets indicate that FODs both effectively capture the essential features governing these properties and have strong interpretability of target attributes. And the remarkably small RMSEs (< 0.1 eV) further confirm the reliability of our predictions at the chemical accuracy level.
To further validate the effectiveness of FODs, we conduct comparative predictions using the molecular orbitals of complete DAD molecules (MODs) as alternative descriptors, which include four key features: HOMO, LUMO, HOMO-1, and LUMO+1 of the intact DADs. Employing the same eight ML models and evaluation protocol, we systematically compare the predictive performance between FODs and MODs. As shown in Figure 4 (radar chart of R2 values; see Supplementary Figure 1 for RMSE results), FODs consistently outperform MOD across eight models, with only two exceptions (MLP and KNN on ΔETT) and with particularly notable advantages in RF and XGBoost - the two most accurate prediction models. This superior performance (higher R2 and lower RMSE) demonstrates that FODs provide more meaningful structural insights and better predictive capability for target properties ΔETT and ΔEST than whole-molecule descriptors, further confirming the value of our fragment-based approach. Although both descriptor types share identical physical interpretations - differing only in their basis (fragment-level vs. whole-molecule) - they yield markedly different predictive outcomes. The superior performance of FODs across most models demonstrates that fragment-derived physicochemical properties can effectively predict whole-molecule characteristics. These results validate the “Fragment-To-Whole” strategy as not only applicable to ML approaches, but potentially advantageous for predictive accuracy in molecular design.
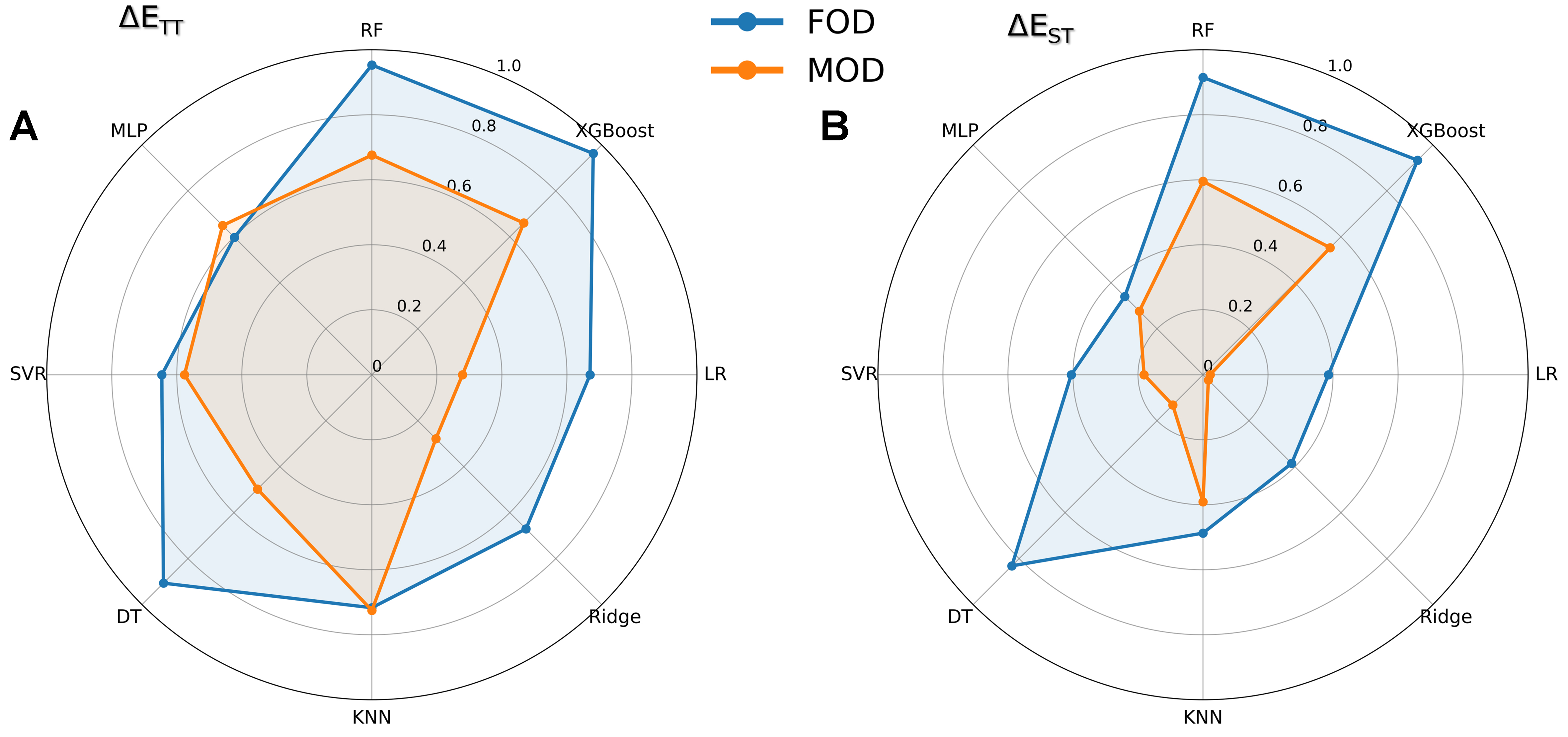
Figure 4. Comparative performance of FODs vs. MODs across eight ML models. Radar plots display R2 values for predictions of (A) ΔETT and (B) ΔEST, where radial distance from the center corresponds to prediction accuracy (greater R2 values extend further toward the outer edge). Each axis represents one ML algorithm, enabling direct visual comparison of descriptor effectiveness. FODs: Fragment orbital descriptors; MODs: molecule orbital descriptors of complete DAD molecules; ML: machine learning; R2: the coefficient of determination; RF: random forest; MLP: multilayer perceptron; SVR: support vector regression; DT: decision tree; KNN: K-nearest neighbors; LR: linear regression; XGBoost: eXtreme gradient boosting.
Therefore, from our results, for DADs, which are typical molecules composed of fragments, it is possible to prioritize focusing on the physicochemical properties of individual molecular fragments when selecting descriptors. This result aligns with the fundamental nonlinear nature of ML, which seeks to establish complex mappings between inputs and outputs, mirroring the nonlinear processes underlying molecular formation. Specifically, when fragments combine to form a complete molecule, their properties undergo nonlinear transformations mediated by quantum mechanical interactions (e.g., electronic coupling, orbital hybridization, and radiative transitions). Fragment-based descriptors may preserve critical electronic and steric information that may be obscured or averaged out in whole-molecule representations, thereby providing ML models with more discriminative features for prediction. Therefore, the “Fragment-To-Whole” approach inherently complements ML’s strength in learning hierarchical, nonlinear relationships to some extent - a task less suited to conventional whole-molecule descriptors that may oversimplify local fragment contributions.
D/A selectivity of energy gaps
Micro-interpretability analysis
The superior predictive performance of FODs for ΔETT and ΔEST using RF and XGBoost stems from their fundamental mechanistic basis. To elucidate this relationship, we employ interpretable ML techniques: (i) PDP and ICE analyses to quantify the marginal effects of individual FOD features on energy level differences; and (ii) SHAP to evaluate the relative contribution of each feature to target predictions. This multifaceted interpretability framework not only enhances our understanding of the quantum mechanical relationships between fragment properties and molecular energy characteristics, but also improves the practical utility of the models for molecular design at the theoretical level.
PDP and ICE analyses share a common theoretical framework for feature effect evaluation while differing in analytical focus. Both methods systematically vary the target feature of interest while holding other features constant to examine the target’s predicted responses. The core distinction emerges in their observational scale: PDP aggregates responses across the entire dataset to reveal average marginal effects, whereas ICE delineates sample-specific response patterns. The two methods’ characteristics are clearly demonstrated in Figures 5 and 6 through XGBoost model visualizations, where red PDP curves represent global trends and blue ICE curves depict individual variations for target attributes ΔETT/ΔEST, respectively. Both RF and XGBoost models show consistent directional trends in PDP and ICE patterns, confirming robust agreement in feature-target relationships. While XGBoost captures finer details via gradient boosting and RF produces smoother curves through bagging [Supplementary Figures 2 and 3], this convergence across the two algorithms validates the reliability of the observed patterns. Meanwhile, as we can see, most ICE curves align well with the PDP trends, with only minor variations observed in a few samples. Therefore, our subsequent analyses focus primarily on the PDP results.
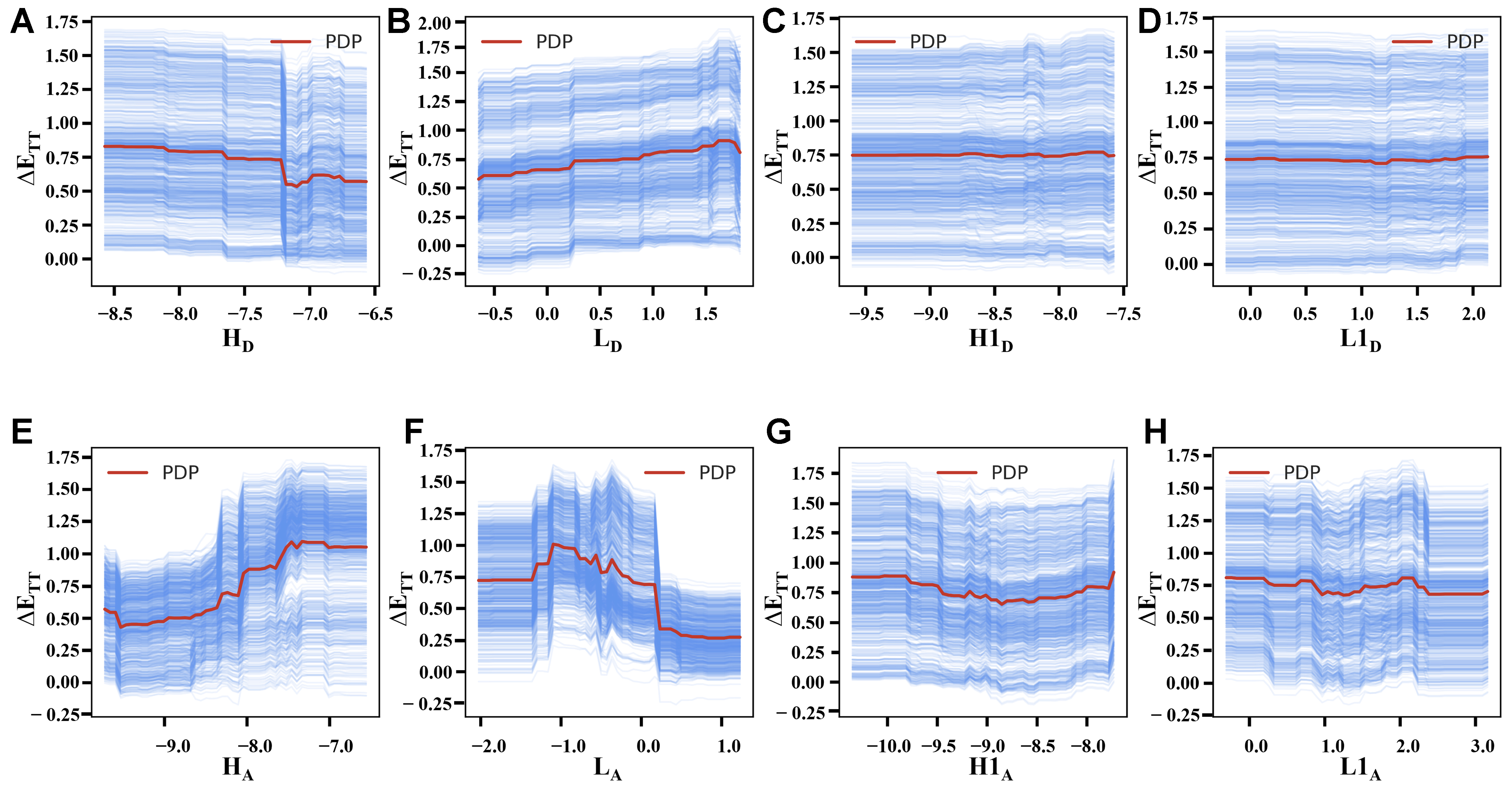
Figure 5. Features’ marginal effect analysis for ΔETT: PDP (red) and ICE (blue) plots of FODs under XGBoost, where (A-H) correspond to features HD, LD, H1D, L1D, HA, LA, H1A, L1A, respectively. PDP: Partial dependence plots; ICE: individual conditional expectation; FODs: fragment orbital descriptors; XGBoost: eXtreme gradient boosting.
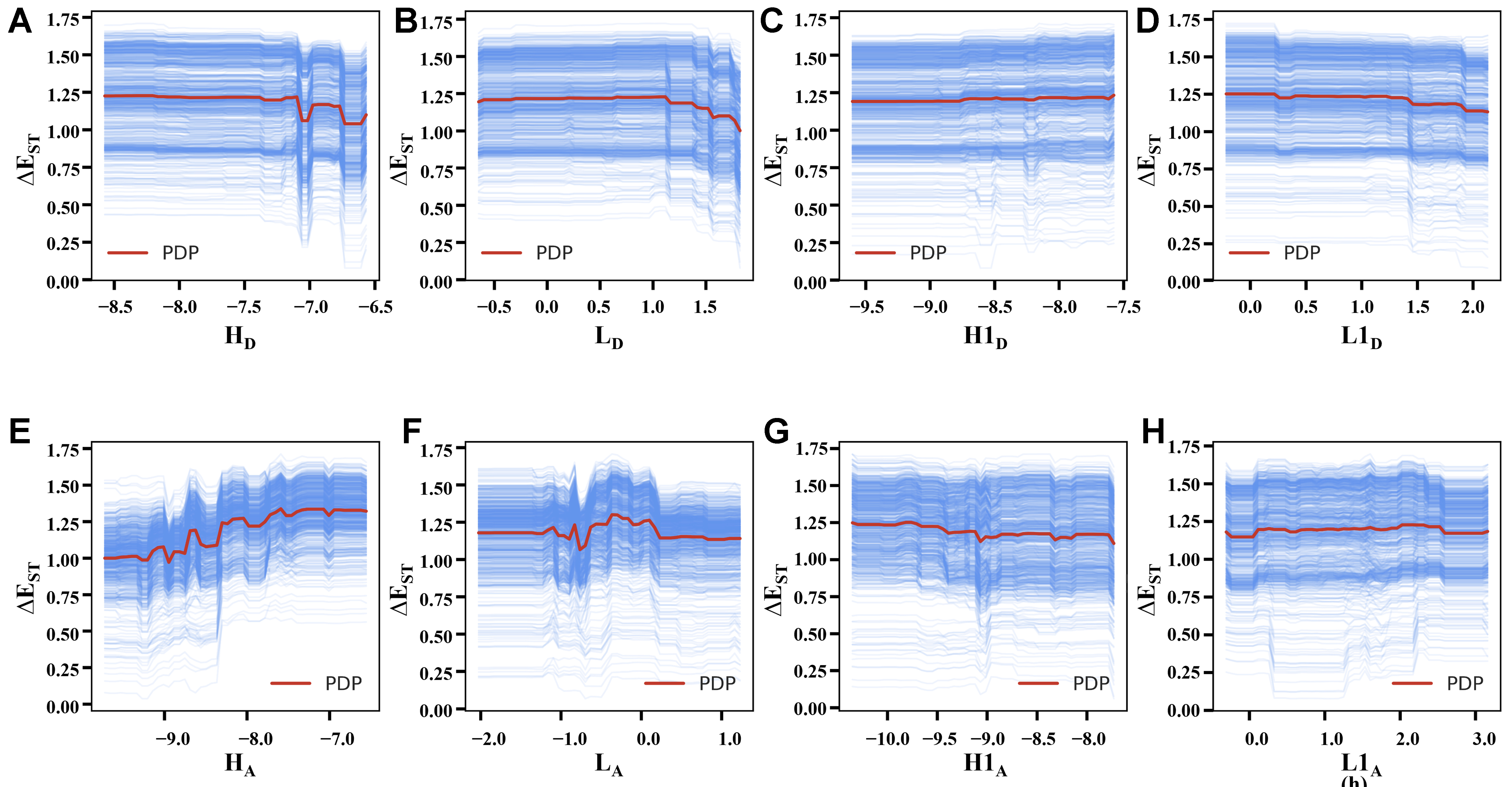
Figure 6. Features’ marginal effect analysis for ΔEST: PDP (red) and ICE (blue) plots of FODs under XGBoost, where (A-H) also correspond to features HD, LD, H1D, L1D, HA, LA, H1A, L1A. PDP: Partial dependence plots; ICE: individual conditional expectation; FODs: fragment orbital descriptors; XGBoost: eXtreme gradient boosting.
The analyses of PDP and ICE charts primarily examine two key characteristics of the curves: (i) the trend of changes, indicating the nature of marginal effects (linear/nonlinear, positive/negative, etc.); and (ii) the intensity of changes, reflected through curve slopes and variation ranges that demonstrate the sensitivity of ΔETT and ΔEST to each FOD - stronger curve fluctuations and wider variation ranges correspond to more significant marginal effects. Therefore, Figure 5 reveals that HA and LA exhibit the most pronounced PDP curve variations, demonstrating their dominant marginal influence of ΔETT. And they also show contrasting nonlinear trends: HA displays a positive correlation in general (higher values are more likely to increase ΔETT), while LA shows a negative correlation (higher values are more likely to decrease ΔETT). Other FODs exhibit weaker nonlinear effects, with H1D and L1D showing minimal influence as evidenced by their near-flat PDP curves (slope ≈ 0). Comparative analyses in Figure 6 show ΔEST responds less dramatically to FODs than ΔETT, with generally smaller PDP curve variations. In the results of ΔEST, although the impact of LA has weakened compared to ΔETT, HA maintains its prominent nonlinear effect, exhibiting a characteristic fluctuating positive correlation and suggesting that increased HA values tend to correlate with enhanced ΔEST values.
While the marginal effects of individual FOD differ between ΔETT and ΔEST, several key similarities emerge. First, both HA and LA consistently demonstrate the strongest marginal impacts among all FODs, with HA exhibiting particularly pronounced effects on these two target attributes. Second, comparative analyses of Figures 5 and 6 reveals that features in the lower rows (acceptor fragments) consistently show greater variation than their upper-row counterparts (donor fragments) with equivalent physical meanings. To establish a comprehensive understanding, we extend this analysis to ΔETS prediction using both RF and XGBoost, which also show excellent predictive performance [Supplementary Figure 4]. The PDP/ICE analyses of ΔETS [Supplementary Figures 5 and 6] also shares the common traits: (i) Significant effects in HA/LA - but LA exhibits stronger marginal effects than HA for ΔETS, contrasting with their relative impacts on ΔETT/ΔEST; (ii) Preservation of the acceptor > donor influence pattern observed in ΔETT/ΔEST systems. These consistent patterns across different targets underscore the pivotal role of acceptor fragments for DADs’ energy gaps, which will also be further validated in subsequent analyses.
While PDP and ICE analyses provide valuable visual assessment of how each FOD influences the target attributes, they lack quantitative measures of feature importance. To address this limitation, we then employ SHAP analysis - a game theory-based interpretability framework that quantifies the marginal contribution of each feature through SHAP values. This strategy not only provides precise quantification of individual FOD’s contributions to ΔETT and ΔEST predictions, but also establishes cross-validation with PDP and ICE results, thereby ensuring comprehensive and robust interpretation of feature impacts. The SHAP method calculates feature importance by determining the weighted average contribution of each descriptor across all possible combinations, providing a mathematically rigorous assessment of feature contributions.
Figure 7 visualizes XGBoost’s SHAP analysis for ΔETT/ΔEST predictions (RF results in Supplementary Figure 7) and Supplementary Figure 8 also shows the results for ΔETS. The vertical arrangement of FODs directly corresponds to their relative impact on energy gap predictions, with the most influential features positioned at the top. Meanwhile, the color gradient of each FOD reflects its correlation with SHAP values. It is worth noting that the positive and negative SHAP values also respectively indicate opposite effects on targets: higher positive SHAP values correspond to greater increases in target values (demonstrating a positive correlation), while negative SHAP values reflect an inverse relationship where descriptors decrease the predicted energy gaps’ values. While XGBoost and RF exhibit discrepancies in their SHAP-based feature importance rankings - particularly for lower-contributing descriptors due to inherent differences in their ensemble methodologies (boosting vs. bagging) - both approaches consistently identify HA and LA as the dominant features. Crucially, the fundamental mechanisms through which these key descriptors influence the energy gaps remain consistent across both models, as evidenced by their identical response patterns in SHAP value distributions (color gradients).
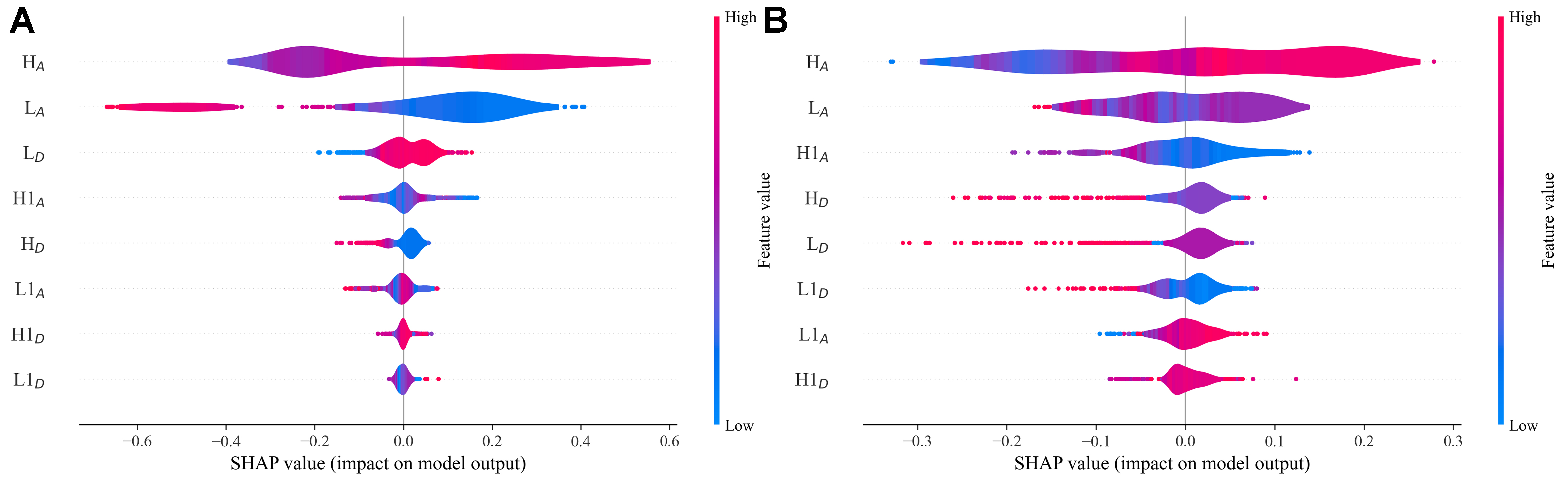
Figure 7. SHAP analysis results for (A) ΔETT and (B) ΔEST predictions using the XGBoost model. The horizontal axes represent SHAP values quantifying each descriptor’s contribution, with positive/negative values indicating enhancing/reducing effects on the target energy gaps. Vertical descriptor ordering reflects their relative importance, while the color gradient (blue to red) encodes the actual values of each FOD, with blue and red corresponding to low and high values, respectively. SHAP: SHapley Additive exPlanations; XGBoost: eXtreme gradient boosting; FODs: fragment orbital descriptors.
And to quantitatively assess the impacts of FODs, Figure 8 and Supplementary Figure 9 present the percentage contribution of each FOD under XGBoost/RF respectively, calculated as the ratio of its SHAP value to the sum of absolute SHAP values across all descriptors. The percentage contribution analysis reaffirms the critical importance of HA and LA as the top two most influential descriptors across all FODs, though their relative impacts vary significantly among different energy gaps: while HA and LA show comparable contributions for ΔETT/ΔETS (within 10% difference), with HA being slightly more dominant for ΔETT and LA prevailing for ΔETS, HA demonstrates clear dominance for ΔEST with approximately twice the contribution of LA. More significantly, the acceptor fragment A collectively accounts for the majority of energy gap determination across all three properties, as consistently shown by both XGBoost and RF models (nearly 77.2%/86.3% for ΔETT, 73.1%/79.2% for ΔEST, and 71.7%/77.5% for ΔETS, respectively). This overwhelming predominance quantitatively validates the central role of acceptor fragment A in governing the electronic structure of DAD systems, highlighting its backbone function in energy gap characterization, which is supported by quantum-mechanical analysis of HLCT states[18], which indicates that the acceptor moiety has a greater impact on the excitation energy levels.
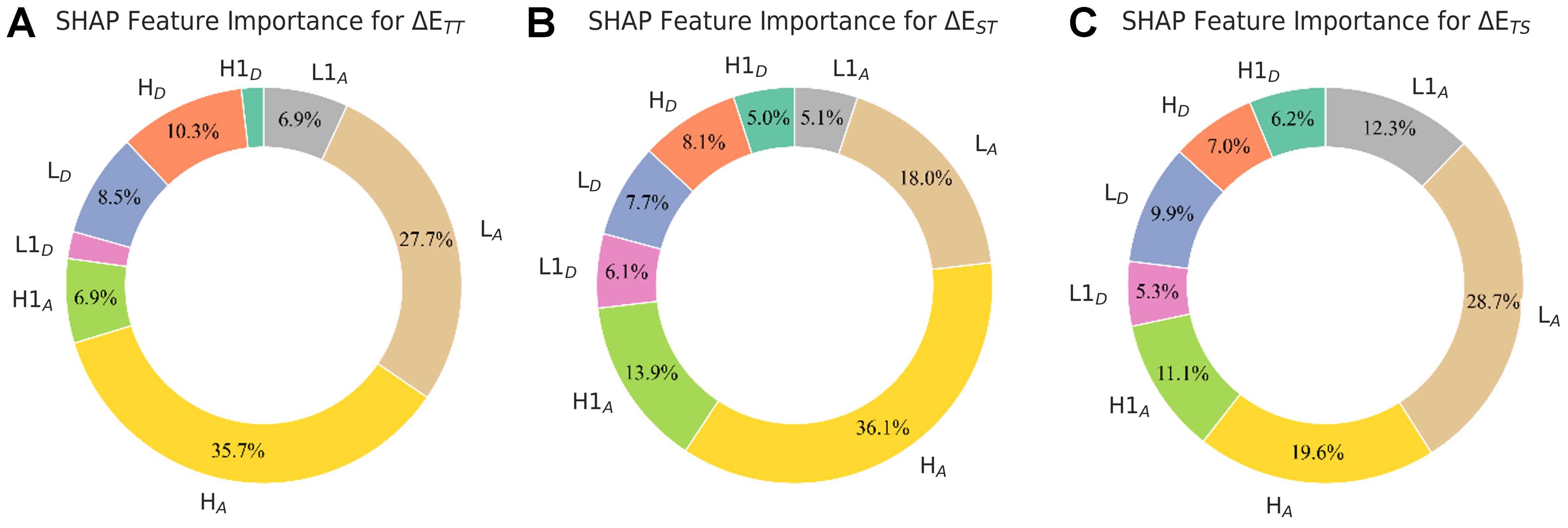
Figure 8. SHAP-derived importance allocations of FODs to energy gap predictions. Circular plots display the percentage distribution of SHAP values for each FOD in XGBoost predictions of (A) ΔETT, (B) ΔEST, and (C) ΔETS. Only features’ contributing > 5% to the prediction are labeled with their percentage values. SHAP: SHapley Additive exPlanations; FODs: fragment orbital descriptors; XGBoost: eXtreme gradient boosting.
Besides, from Figure 7 we can see that HA exhibits a consistent positive influence on all three target energy gaps, as evidenced by the progressive color shift from blue (low values) to red (high values) corresponding to increasing SHAP values. In contrast, LA demonstrates a more complex behavior - showing strong negative correlations with ΔETT and ΔETS, while displaying a non-monotonic relationship with ΔEST. The relationships between the remaining features and the SHAP values of target properties are more complex. All of these trends are further quantified in Supplementary Figures 10-12, which plot SHAP values against actual FOD values under XGBoost. The scatterplots not only refine the color gradient patterns observed in Figure 7, but also show general consistency with corresponding PDP trends. This agreement between SHAP and PDP analyses - despite their different methodological approaches - provides mutual validation of the identified feature-property relationships.
Macro-statistical analysis
The micro-interpretability analysis reveals complex, nonlinear relationships between DADs’ energy gaps and each FOD with varying marginal effects and contributions. While FODs quantify fragment-level electronic characteristics, the macroscopic combination of D/A fragments generates synergistic effects that collectively combine or transcend individual descriptor contributions. This suggests potential fragment selectivity in energy gap modulation - specific D/A combinations may preferentially yield DAD molecules with particular energy-gap value ranges. We subsequently validate this hypothesis through macro-statistical analysis.
Based on ML predictions for 5,400 DAD molecules, we analyze the fragments’ composition of two strategically selected subsets: (i) the top 500 molecules with the largest predicted ΔETT values (inhibiting the IC process); and (ii) the top 500 molecules with the smallest predicted ΔEST values (favoring the RISC process). Figure 9 displays pie charts illustrating the proportional distribution of D/A fragments and their substituents in those high-performance subsets (with absolute occurrence counts provided in Supplementary Figures 13 and 14).
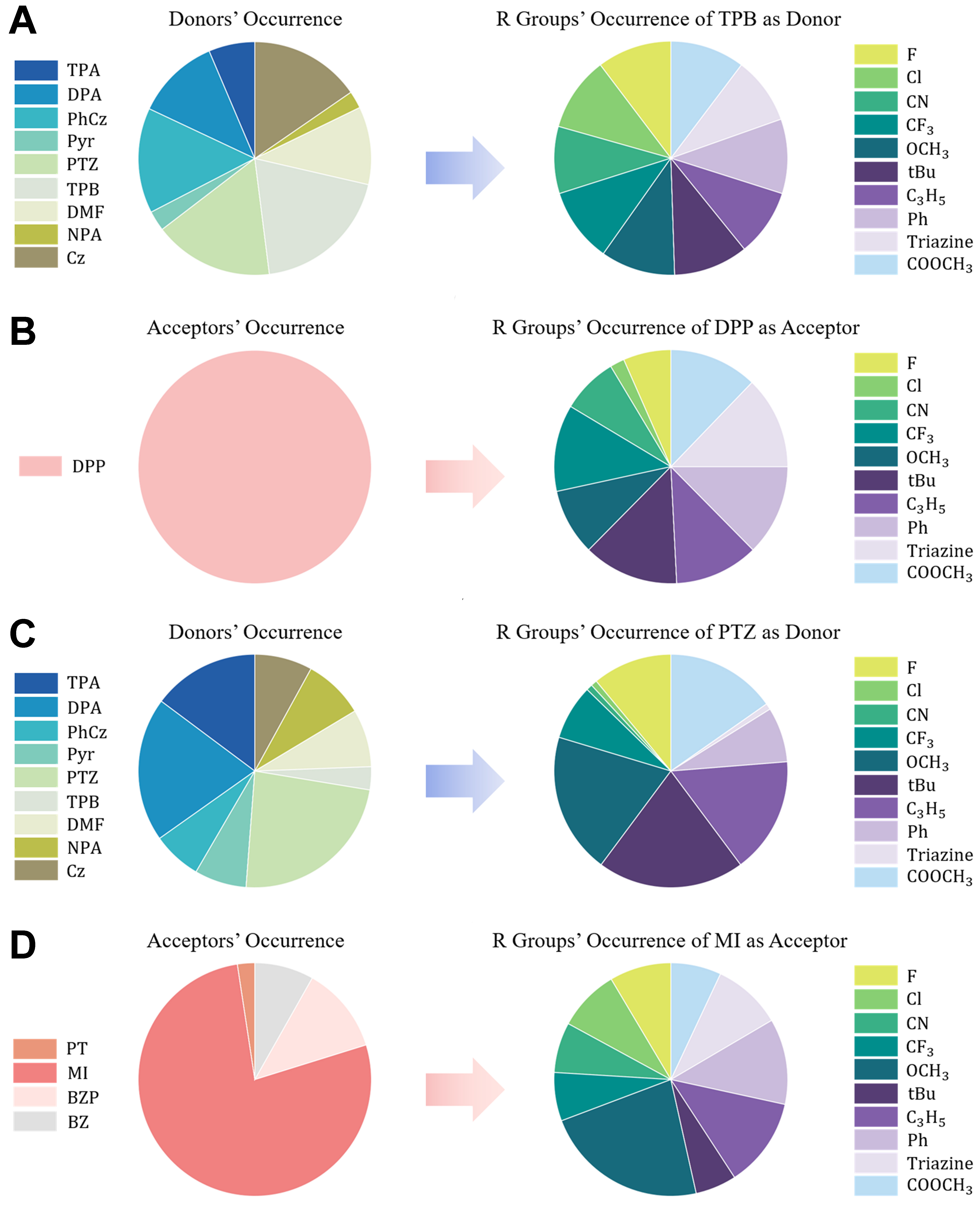
Figure 9. Compositional analysis of D/A fragments for ΔETT and ΔEST subpopulations: (A) donor distribution and (B) acceptor distribution with representative substituents (TPB and DPP, repectively) for top 500 large-ΔETT DADs; (C and D) Equivalent distributions for top 500 small-ΔEST performers (PTZ/MI-dominated). D/A: Donor/acceptor; DADs: donor–acceptor–donor triads.
The frequency distributions of D/A fragments exhibit distinct patterns between the two energy-gap scenarios, with particularly marked differences in acceptor fragments (A). Strikingly, DPP appears exclusively in the top 500 large-ΔETT DADs, while MI dominates (77.4%) the small-ΔEST subset. Donor fragments (D) exhibit relatively balanced but slight distinct distributions, with TPB prevailing in large-ΔETT systems and PTZ predominating in small-ΔEST configurations. A similar trend is also observed for ΔETS: analysis of the top 500 molecules with largest ΔETS values (using large-ΔETT based on the energy-gap relationship ΔETS = ΔETT - ΔEST) reveals significant variability in fragment proportion distributions, particularly for acceptor fragments, as shown in Supplementary Figure 15. And the large-ΔETS selects not only the MI fragment selected by the small-ΔEST, but also the DPP fragment selected by the large-ΔETT (it will be seen further on that the Ant fragment selected by large-ΔETS is also selectable by large-ΔETT). The substituents also exhibit non-complete-uniform distribution patterns (particularly for ΔEST systems), indicating their significant role in modulating D/A fragment properties. The FODs values of the donor and acceptor changed upon substitution, illustrating that substituent effects are primarily encoded indirectly via shifts in FOD values and contribute to the predictions.
These statistical trends demonstrate clear fragment specificity - different D/A fragments preferentially yield distinct energy gap characteristics (or specific energy gap values exhibit selective preferences for particular D/A fragments). This macroscopic selectivity originates from fundamental microscopic mechanisms: variations in FOD feature distributions across fragments create unique electronic environments, where differential marginal effects and contributions from individual features collectively determine the final energy gaps.
To further elucidate the fragments’ selectivity, we analyze the joint distribution of predicted ΔETT and ΔEST values for all 5,400 DAD molecules, categorized by D/A fragment combinations [Figure 10]. The result reveals striking spatial patterns: acceptor (A) fragments exhibit tightly clustered distributions, whereas donor (D) fragments show broader dispersion with limited localized heterogeneity. This demonstrates strong cooperative selectivity of ΔETT and ΔEST for acceptor fragments. And these findings directly inform rational DAD design strategies: (i) Acceptor-driven control: the acceptor type primarily determines the ΔETT/ΔEST ranges (e.g., DPP/Ant for large ΔETT; MI for small ΔEST, which has been supported by relevant literature[44-51]); (ii) Donor fine-tuning: donor selection provides secondary modulation within these acceptor-defined ranges. For systems requiring simultaneous optimization of both energy gaps, the fragment combination space can be also navigated using this hierarchical design principle - first selecting acceptors to establish the target energy gap ranges, then choosing donors to refine the electronic properties.
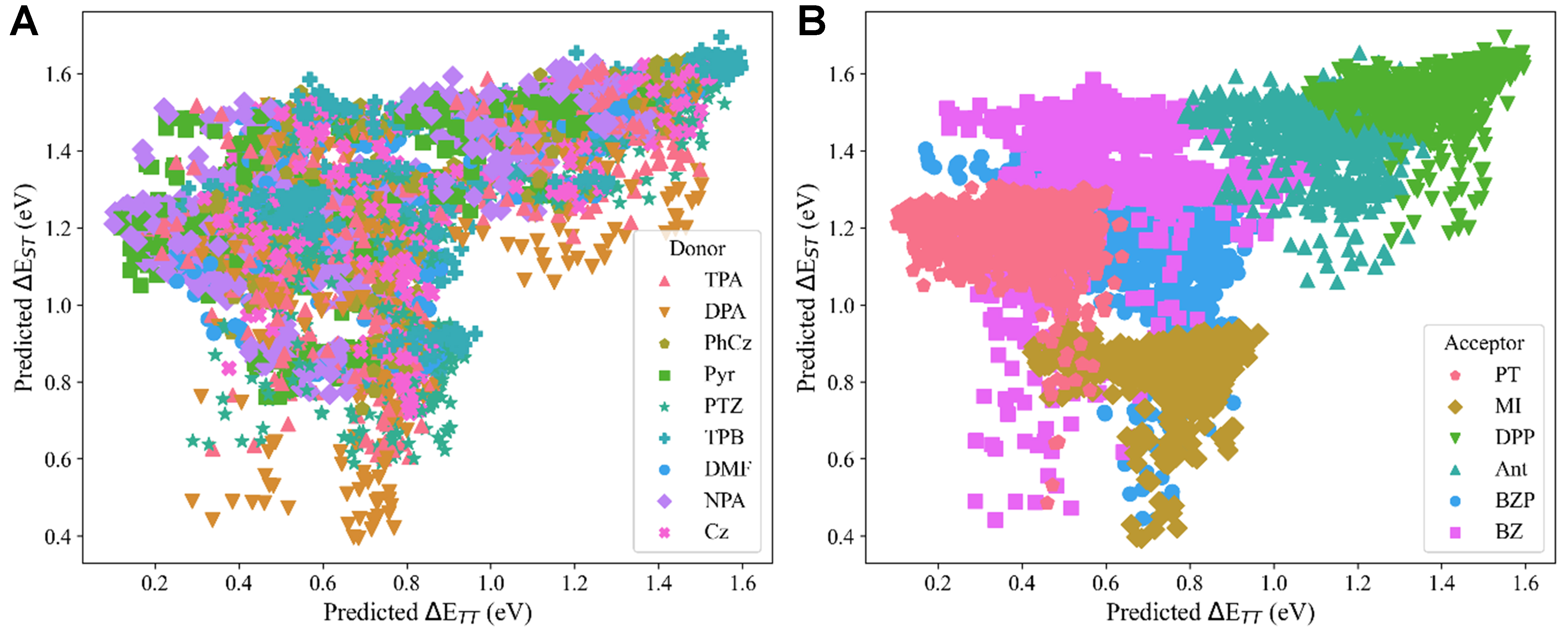
Figure 10. Correlation between predicted ΔETT and ΔEST values for 5,400 DAD molecules, visualized by (A) donor fragments and (B) acceptor fragments. Color-coding represents different fragment categories, revealing distinct clustering patterns between donor and acceptor contributions to energy-gap modulation. DAD: Donor–acceptor–donor.
The observed fragments’ selectivity in DAD molecular energy gaps provides fundamental insights into whole-molecule/fragment relationships. Crucially, FODs associated with acceptor fragment A exhibit significantly greater marginal effects and average contributions than those of donor fragment D, explaining the dominant macroscopic influence of acceptor fragments. This phenomenon originates from the unique ternary DAD architecture, where the central acceptor mediates strong electronic coupling between the two flanking donors. This coupling enhances cooperative charge transfer from both donors to the acceptor site, thereby amplifying the acceptor’s determining role in energy gap formation.
CONCLUSIONS
In summary, we have developed a robust machine-learning-driven strategy employing FODs to accurately predict critical energy gaps (ΔETT, ΔEST, and ΔETS) within DAD triads, a key class of hot-exciton materials for OLEDs. By adopting a fragment-to-whole predictive framework, our models (particularly RF and XGBoost) demonstrated outstanding predictive accuracy and chemical interpretability, significantly outperforming conventional whole-molecule descriptors. Comprehensive interpretability analyses (PDP, ICE, and SHAP) highlighted the pivotal roles of acceptor fragment orbitals (HA, LA) in determining energy gaps, underlining a pronounced acceptor-driven selectivity pattern. Macro-statistical analysis further validated these microscopic observations, revealing distinct fragment combinations - specifically DPP-based acceptors for large ΔETT and MI-based acceptors for small ΔEST - that consistently achieve desired electronic characteristics. Consequently, we established a hierarchical design principle prioritizing acceptor selection to control primary electronic properties, supplemented by donor fine-tuning for optimal performance. Beyond the DAD triads studied herein, the proposed fragment-centric informatics framework serves as a general paradigm that can be extended to other fragment-assembled architectures, such as D–A copolymers and semiconductors systems, facilitating the high-throughput discovery of functional optoelectronic materials. Additionally, this work substantially advances fundamental understanding of structure-property relationships in OLED materials, providing a practical, data-driven framework for accelerating molecular discovery in organic optoelectronics.
DECLARATIONS
Authors’ contributions
Wrote the main manuscript and prepared the figures: Zhang, H.; Zhang, X.; Zhu, Y.
Prepared molecular code for database: Wang, S.
Optimized the candidates molecules for the validation: Guo, Z.; Zhou, A.
Supervised the project: Meng, H.; Zhu, Y.
All authors reviewed the manuscript.
Availability of data and materials
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by Grants: the National Natural Science Foundation of China (12404460, 22275003, 52573188), Shenzhen Science and Technology Program (JCYJ20241202130509013), GuangDong Engineering Technology Research Center of Multi-Dimensional Optoelectronic Materials(2024B195), Guangdong Key Laboratory of Flexible Optoelectronic Materials and Devices, and the Guangdong Basic and Applied Basic Research Foundation (2023A1515111072, 2025A1515011373).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Jordan, M. I.; Mitchell, T. M. Machine learning: trends, perspectives, and prospects. Science 2015, 349, 255-60.
2. Qiao, X.; Liu, Z.; Sun, J.; et al. Resistive switching oxides: mechanism, performance, and device-algorithm co-design for artificial intelligence. Adv. Mater. 2026, e17373.
3. Feng, B.; Wang, B.; Lv, L.; et al. Interpreting X-ray diffraction patterns of metal-organic frameworks via generative artificial intelligence. J. Am. Chem. Soc. 2026, 148, 869-78.
4. Yu, Y.; Xie, Z.; Luo, M.; et al. Optimizing toward discovery: AI-driven exploration of Lewis acid-base catalysts for PET glycolysis. J. Am. Chem. Soc. 2026, 148, 4635-44.
5. Bin Faheem, A.; Han, Z.; Wu, D.; Li, H. AI-driven big data frameworks for electrode-electrolyte interphases in batteries. Adv. Mater. 2026, 38, e21975.
6. Agrawal, A.; Choudhary, A. Perspective: Materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL. Mater. 2016, 4, 053208.
7. Jose, R.; Ramakrishna, S. Materials 4.0: materials big data enabled materials discovery. Appl. Mater. Today. 2018, 10, 127-32.
8. Pilania, G. Machine learning in materials science: from explainable predictions to autonomous design. Comput. Mater. Sci. 2021, 193, 110360.
9. Stefańska, M.; Müntener, T.; Hiller, S. Predictions of steady-state photo-CIDNP enhancement by machine learning. J. Am. Chem. Soc. 2025, 147, 27172-8.
10. Lu, S.; Zhou, Q.; Ouyang, Y.; Guo, Y.; Li, Q.; Wang, J. Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning. Nat. Commun. 2018, 9, 3405.
11. Ali, A.; Park, H.; Mall, R.; et al. Machine learning accelerated recovery of the cubic structure in mixed-cation perovskite thin films. Chem. Mater. 2020, 32, 2998-3006.
12. Lee, S. Y.; Byeon, S.; Kim, H. S.; Jin, H.; Lee, S. Deep learning-based phase prediction of high-entropy alloys: optimization, generation, and explanation. Mater. Design. 2021, 197, 109260.
13. Li, Y.; Guo, W. Machine-learning model for predicting phase formations of high-entropy alloys. Phys. Rev. Mater. 2019, 3, 095005.
14. Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Hirzel, T. D.; et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 2016, 15, 1120-7.
15. Cheng, Z.; Liu, J.; Jiang, T.; et al. Automatic screen‐out of Ir(III) complex emitters by combined machine learning and computational analysis. Adv. Opt. Mater. 2023, 11, 2301093.
16. Jeong, M.; Joung, J. F.; Hwang, J.; et al. Deep learning for development of organic optoelectronic devices: efficient prescreening of hosts and emitters in deep-blue fluorescent OLEDs. npj. Comput. Mater. 2022, 8, 834.
17. Choi, I.; Amin, A.; Katware, A.; Kang, S. W.; Lee, J. Machine learning algorithm for artificial intelligence-based precise structural modeling in organic light-emitting diodes. ACS. Photonics. 2024, 11, 2938-45.
18. Xu, Y.; Xu, P.; Hu, D.; Ma, Y. Recent progress in hot exciton materials for organic light-emitting diodes. Chem. Soc. Rev. 2021, 50, 1030-69.
19. Li, W.; Pan, Y.; Yao, L.; et al. A hybridized local and charge‐transfer excited state for highly efficient fluorescent OLEDs: molecular design, spectral character, and full exciton utilization. Adv. Opt. Mater. 2014, 2, 892-901.
20. Yang, L.; Kim, V.; Lian, Y.; Zhao, B.; Di, D. High-efficiency dual-dopant polymer light-emitting diodes with ultrafast inter-fluorophore energy transfer. Joule 2019, 3, 2381-9.
21. Liu, B.; Yu, Z. W.; He, D.; et al. Ambipolar D–A type bifunctional materials with hybridized local and charge-transfer excited state for high performance electroluminescence with EQE of 7.20% and CIEy ~ 0.06. J. Mater. Chem. C. 2017, 5, 5402-10.
22. Li, W.; Pan, Y.; Xiao, R.; et al. Employing ~100% excitons in OLEDs by utilizing a fluorescent molecule with hybridized local and charge‐transfer excited state. Adv. Funct. Mater. 2014, 24, 1609-14.
23. Liu, B.; Yuan, Y.; He, D.; et al. High-performance blue OLEDs based on phenanthroimidazole emitters via substitutions at the C6- and C9-positions for improving exciton utilization. Chemistry 2016, 22, 12130-7.
24. Tang, X.; Bai, Q.; Peng, Q.; et al. Efficient deep blue electroluminescence with an external quantum efficiency of 6.8% and CIEy < 0.08 based on a phenanthroimidazole–sulfone hybrid donor–acceptor molecule. Chem. Mater. 2015, 27, 7050-7.
25. Zhu, Y.; Vela, S.; Meng, H.; Corminboeuf, C.; Fumanal, M. Donor–acceptor–donor “hot exciton” triads for high reverse intersystem crossing in OLEDs. Adv. Opt. Mater. 2022, 10, 2200509.
26. Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday. Soc. 1950, 9, 14-9.
27. Chen, T.; Zheng, L.; Yuan, J.; et al. Understanding the control of singlet-triplet splitting for organic exciton manipulating: a combined theoretical and experimental approach. Sci. Rep. 2015, 5, 10923.
28. Froitzheim, T.; Grimme, S.; Mewes, J. M. Either accurate singlet-triplet gaps or excited-state structures: testing and understanding the performance of TD-DFT for TADF emitters. J. Chem. Theory. Comput. 2022, 18, 7702-13.
29. Diesing, S.; Zhang, L.; Zysman-Colman, E.; Samuel, I. D. W. A figure of merit for efficiency roll-off in TADF-based organic LEDs. Nature 2024, 627, 747-53.
30. Huang, S.; Zhang, Q.; Shiota, Y.; et al. Computational prediction for singlet- and triplet-transition energies of charge-transfer compounds. J. Chem. Theory. Comput. 2013, 9, 3872-7.
31. Ju, C. W.; Bai, H.; Li, B.; Liu, R. Machine learning enables highly accurate predictions of photophysical properties of organic fluorescent materials: emission wavelengths and quantum yields. J. Chem. Inf. Model. 2021, 61, 1053-65.
32. Xu, S.; Liu, X.; Cai, P.; Li, J.; Wang, X.; Liu, B. Machine-learning-assisted accurate prediction of molecular optical properties upon aggregation. Adv. Sci. 2022, 9, e2101074.
33. Sun, W.; Zheng, Y.; Yang, K.; et al. Machine learning-assisted molecular design and efficiency prediction for high-performance organic photovoltaic materials. Sci. Adv. 2019, 5, eaay4275.
34. Mao, Y.; Yao, X.; Yu, Z.; An, Z.; Ma, H. Ground-state orbital descriptors for accelerated development of organic room-temperature phosphorescent materials. Angew. Chem. Int. Ed. Engl. 2024, 63, e202318836.
35. Zhang, X.; Ding, B.; Wang, Y.; et al. Machine learning for screening small molecules as passivation materials for enhanced perovskite solar cells. Adv. Funct. Mater. 2024, 34, 2314529.
36. Woon, K. L.; Chong, Z. X.; Ariffin, A.; Chan, C. S. Relating molecular descriptors to frontier orbital energy levels, singlet and triplet excited states of fused tricyclics using machine learning. J. Mol. Graph. Model. 2021, 105, 107891.
37. Sifain, A. E.; Lystrom, L.; Messerly, R. A.; et al. Predicting phosphorescence energies and inferring wavefunction localization with machine learning. Chem. Sci. 2021, 12, 10207-17.
38. Pang, Y.; Peng, Q. Molecular descriptors of excited-state property for high-throughput screening of organic photofunctional materials. J. Phys. Chem. Lett. 2024, 15, 8804-12.
39. He, Z.; Bi, H.; Liang, B.; Li, Z.; Zhang, H.; Wang, Y. Frontier molecular orbital weighted model based networks for revealing organic delayed fluorescence efficiency. Light. Sci. Appl. 2025, 14, 75.
40. Deng, J.; Liang, J.; Bai, S. Learning intermolecular electronic coupling with molecular-orbital-based descriptors. J. Phys. Chem. Lett. 2024, 15, 12551-60.
41. Comas-Vilà, G.; Salvador, P. Quantification of the donor-acceptor character of ligands by the effective fragment orbitals. Chemphyschem 2024, 25, e202400582.
42. Zeng, S.; Zhao, Y.; Li, G.; Wang, R.; Wang, X.; Ni, J. Atom table convolutional neural networks for an accurate prediction of compounds properties. npj. Comput. Mater. 2019, 5, 223.
43. Blaskovits, J. T.; Fumanal, M.; Vela, S.; Corminboeuf, C. Designing singlet fission candidates from donor–acceptor copolymers. Chem. Mater. 2020, 32, 6515-24.
44. Shan, X.; Lu, X.; Sahoo, S. R.; et al. Bright long-wavelength multi-state TADF emitters. Angew. Chem. Int. Ed. Engl. 2025, 64, e202507793.
45. Fan, T.; Nie, X.; Feng, S.; et al. Efficient circularly polarized near-infrared hybridized local and charge-transfer emitter D-A-D structures with TPA as donor and a chiral (R/S) alkyl-substituted thiadiazolo[3,4-g]quinoxaline. Chem. Eng. J. 2025, 525, 170368.
46. Wang, R.; Hu, T.; Liu, Y.; et al. Highly efficient, red delayed fluorescent emitters with exothermic reverse intersystem crossing via hot excited triplet states. J. Phys. Chem. C. 2020, 124, 20816-26.
47. Chen, X.; Yang, Z.; Li, W.; et al. Nondoped red fluorophores with hybridized local and charge-transfer state for high-performance fluorescent white organic light-emitting diodes. ACS. Appl. Mater. Interfaces. 2019, 11, 39026-34.
48. Pun, A. B.; Campos, L. M.; Congreve, D. N. Tunable emission from triplet fusion upconversion in diketopyrrolopyrroles. J. Am. Chem. Soc. 2019, 141, 3777-81.
49. Naimovicius, L.; Wolek, L.; Zhang, S. K.; Kim, J. E.; Tauber, M. J.; Pun, A. B. Activating solid-state triplet-triplet annihilation upconversion via bulky annihilators. J. Am. Chem. Soc. 2026, 148, 3811-9.
50. Feng, H. J.; Zhang, M. Y.; Jiang, L. H.; Huang, L.; Pang, D. W. Triplet-triplet annihilation upconversion: from molecules to materials. Acc. Chem. Res. 2025, 58, 3543-57.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.