


Machine learning-assisted high-entropy alloy discovery: a perspective
 0
0 Abstract
High-entropy alloys (HEAs) have attracted extensive attention due to their exceptional mechanical, physical, and chemical properties, making them promising candidates for extreme environments. Understanding the complex structure-property relationships in these multi-principal element systems is crucial for discovering and designing high-performance HEAs. However, their vast compositional space and high-dimensional chemical complexity pose major challenges to traditional trial-and-error design. Machine learning (ML) offers a transformative strategy to overcome these barriers by enabling data-driven exploration. This perspective first reviews the critical challenges currently limiting HEA development, then summarizes recent ML breakthroughs in phase formation prediction, multi-objective optimization, and accelerated atomistic simulations. Finally, we discuss ongoing challenges and propose future opportunities for integrating ML with experimental and computational methods to create more interpretable, data-efficient, and autonomous ML-driven HEA design frameworks.
Keywords
INTRODUCTION
The discovery and optimization of advanced alloys have traditionally relied on empirical knowledge and computationally expensive simulations. This conventional trial-and-error approach is particularly limiting for high-entropy alloys (HEAs) due to their vast compositional space and complex microstructures[1-4]. HEAs exhibit outstanding mechanical strength[5,6], superior radiation tolerance[7], remarkable corrosion and oxidation resistance[8-10], making them promising candidates for demanding applications in aerospace, nuclear reactors, and other extreme environments. However, the exploration of high-performance HEAs often requires extensive experimentation and high-throughput calculations[11,12], making rapid innovation difficult.
Recent advances in machine learning (ML) have introduced a transformative shift in materials design, often referred to as the “fourth paradigm” of scientific discovery[13]. Unlike the first three paradigms—experimental science, theoretical modeling, and computational simulation[14]—the fourth paradigm leverages data-driven strategies to autonomously learn from existing knowledge and predict material behavior without relying on explicit physical models[15]. ML enables accelerated exploration of vast compositional spaces, efficient optimization of mechanical properties, and the discovery of unconventional alloy chemistries that might be overlooked by human intuition or traditional approaches[16,17]. Despite this potential, ML applications in HEA research still face critical challenges, such as model opacity, data scarcity, and poor interpretability[18,19].
This perspective highlights how ML is redefining the design paradigm of high-performance HEAs. We begin by discussing the current limitations in HEA design. Then, we briefly summarize the recent successful applications of ML in HEAs. Finally, we explore the challenges and opportunities in applying ML to the design and discovery of HEAs and provide some insights into the future development of this field.
LIMITATIONS OF CONVENTIONAL HEA DESIGN
Despite extensive studies on HEAs over the past decade due to their outstanding properties, significant challenges still remain in their design and optimization. (1) Composition-structure-property relationships. Due to their chemically disordered nature, HEAs are not restricted to equiatomic compositions[20]. Numerous potential elements, particularly transition metals, can be incorporated, resulting in a nearly limitless compositional space. Efficiently exploring this vast space, fine-tuning alloys for targeted properties, and uncovering the underlying correlations within the complex composition-structure-property database present formidable challenges; (2) Multi-objective optimization. Designing high-performance alloys often requires simultaneous optimization of multiple, sometimes conflicting properties—such as strength-ductility[21,22], high hardness and corrosion resistance with low density[23], or oxidation resistance combined with excellent fracture toughness[24]. The complex and nonlinear trade-offs between these properties, make it exceedingly difficult to identify Pareto-optimal solutions using conventional trial-and-error or single-objective approaches. (3) Theoretical modeling costs. Modeling HEAs is hindered by numerous chemical interactions and high computational costs of first-principles simulations for large disordered supercells[25].
ML-DRIVEN BREAKTHROUGHS IN HEA RESEARCH
Designing for extrapolation in sparse HEA regimes
Most ML models for HEAs achieve strong performance inside well-sampled compositional regions but often fail to generalize reliably to sparsely sampled or novel chemical spaces[26]. Addressing this requires an extrapolation-first design strategy that couples rigorous uncertainty quantification (UQ), and active-data acquisition strategies[27]. For example, Yang et al. applied a two-step generative adversarial network-based augmentation pipeline to generate realistic synthetic samples and enrich a limited hardness dataset[28]; by expanding the diversity of input compositions and descriptors, they demonstrated improved model robustness and out-of-domain predictive performance for underrepresented compositions. Sulley et al.[29] implemented an active-learning workflow driven by a neural-network surrogate and Bayesian optimization: the surrogate identifies high-uncertainty candidates, the acquisition function balances exploration and exploitation, and selected compositions are evaluated with high-fidelity calculations to iteratively update the model—this procedure substantially reduces the number of expensive evaluations needed to locate promising regions. Together, these studies show that combining data augmentation, UQ-aware model selection, and active sampling is a practical route to trustworthy extrapolation across the vast HEA design landscape.
Phase formation prediction
Traditional approaches, such as empirical phase formation rules[30,31], CALPHAD[32,33] modeling, and density functional theory (DFT)-based thermodynamic calculations[34,35], face challenges due to high computational demands. ML has significantly advanced phase formation prediction by learning complex nonlinear relationships between alloy composition and phase stability[36]. For instance, neural networks have been employed to improve the prediction accuracy of amorphous, intermetallic and solid solution phases[37]. Figure 1A highlights an ML framework introduced by Zhou et al. that refines classic phase design rules[37]. Three ML algorithms were trained on 13 thermodynamic and electronic descriptors, achieving testing accuracies above 95% for distinguishing amorphous, intermetallic and solid solution phases. Using the artificial neural network algorithm shown in Figure 1A, the authors derived a sensitivity matrix from the ML model. And this matrix allowed for a quantitative assessment of how to adjust a design parameter to achieve the formation of a specific phase, as shown in Figure 1B. However, in these approaches, nonlinear algorithms often function as “black boxes”, making it difficult to interpret the relationship between inputs and outputs.
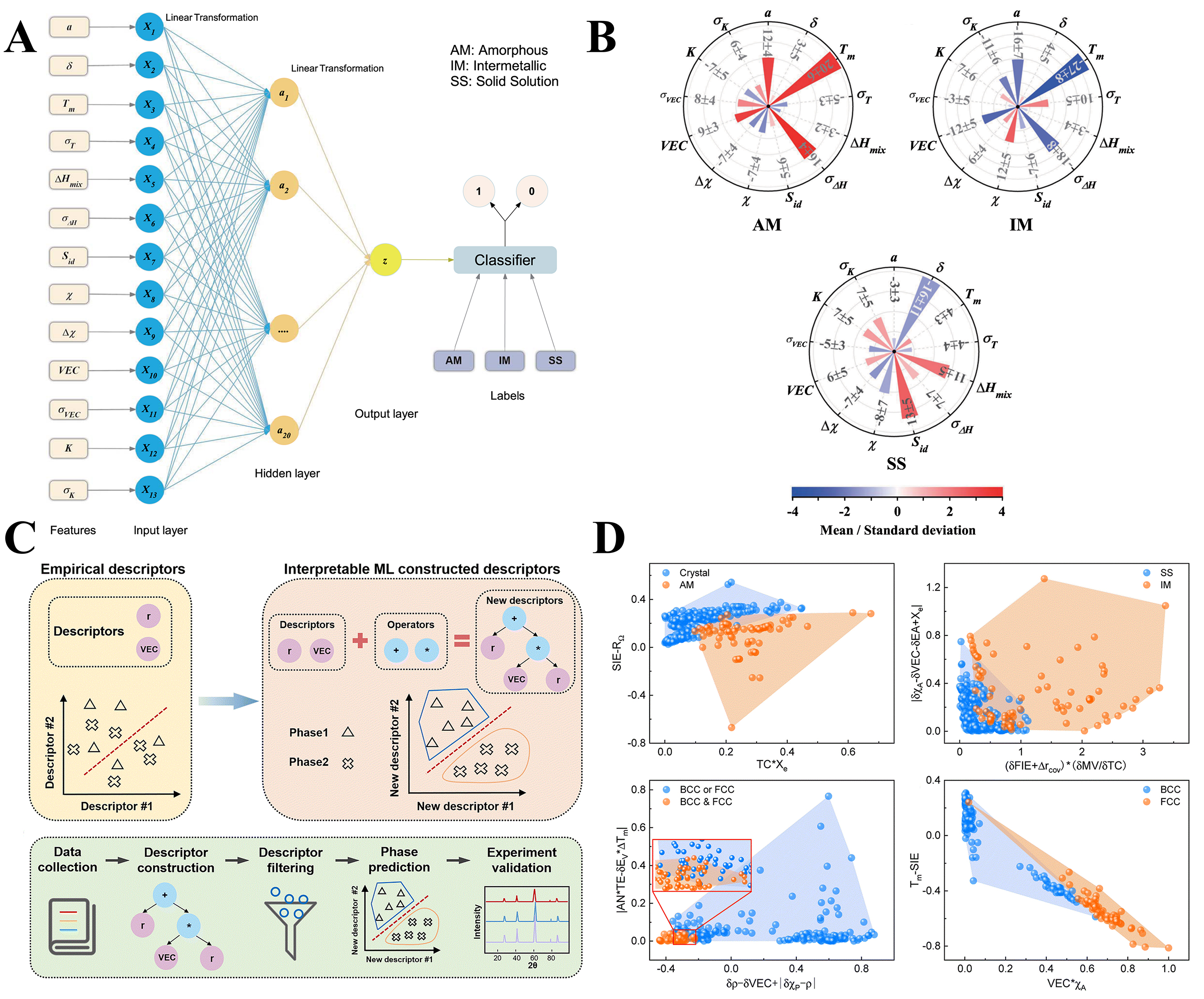
Figure 1. Applications of ML in predicting phase formation of HEAs. (A) Schematic of the artificial neural network-based ML model; (B) Comparison of the sensitivity measures of the 13 design parameters based on the result of the ANN model. The figures are quoted with permission from Zhou et al.[37]; (C) Prediction based on empirical descriptors and interpretable ML, and the workflow for obtaining robust descriptors using interpretable ML algorithms; (D) The best 2D descriptors for phase prediction and subfigures represent the result for four categories: amorphous and crystal; solid solution and intermetallic; single BCC or FCC and dual BCC & FCC phases; and FCC and BCC. The figures are quoted with permission from Zhao et al.[38].
Zhao et al. employed the interpretable ML algorithm SISSO (Sure Independence Screening and Sparsifying Operator) to develop novel descriptors for phase prediction in HEAs[38], and the procedure for constructing new descriptors for phase prediction is shown in Figure 1C. Figure 1D shows the two-dimensional (2D) descriptor based 2D maps for four categories. In each figure, two convex hulls with different colors represent different phases. The overlap between the two convex hulls indicates that the datapoints are difficult to distinguish. This interpretable model achieved approximately 75% accuracy in distinguishing solid solution from intermetallic phases and around 95% in differentiating face-centered-cubic (FCC) from body-centered-cubic (BCC) phases. Experimental synthesis of 25 additional alloys validated the robustness of these descriptors and provided mechanistic insights into phase stability[38].
Collectively, these studies demonstrate how different classes of ML models complement one another in advancing phase-formation prediction. A concise comparison of their datasets, model architectures, and performance metrics is summarized in Table 1.
A brief overview of ML approaches discussed above for HEA phase formation prediction
| Study | Algorithm | Target | Features | Dataset size (train/val/test) | Accuracy | Elementally OOD |
| Ref.[37] | ANN | AM | 13 | 601 (70/15/15%) | 98.9% | No |
| SS | 97.8%, | |||||
| IM | 95.6% | |||||
| CNN | AM | 97.8% | ||||
| SS | 98.9% | |||||
| IM | 94.4% | |||||
| SVM | AM | 96.7% | ||||
| SS | 98.9% | |||||
| IM | 95.6% | |||||
| Ref.[38] | SISSO | SS or IM | 85 | 541 (80/0/20%) | ~75% | No |
| BCC or FCC BCC & FCC | ~79% | |||||
| crystal or AM | ~95% | |||||
| BCC or FCC | ~95% |
Multi-objective optimization of mechanical properties
Designing HEAs with optimal combinations of mechanical properties poses a formidable challenge due to inherent property trade-offs. For instance, lightweight refractory high-entropy alloys (LW-RHEAs) are anticipated to exhibit a unique combination of low density, high strength, high hardness, and excellent corrosion resistance[39,40]. To balance the often conflicting constraints among these four key mechanical properties and phase structures, Gao et al. employed a sequential filtering strategy to design Al-Nb-Ti-V-Cr-Mo-based LW-RHEAs[41], as shown in Figure 2A. By integrating ML models for phase prediction and hardness estimation with interpretable feature selection—such as utilizing the SHapley Additive exPlanations (SHAP) method—the authors screened nearly one million candidate compositions and filtered them layer by layer using property thresholds. Constraints on the single-phase bcc_A2 structure, hardness, Cr content, low density, and high melting point were used to refine the design space, as shown in Figure 2B[41]. The final three designed alloys achieved excellent combinations of hardness and corrosion resistance, with experimental validation confirming the predicted properties and phase stability. While effective, this one-shot design framework lacks adaptive feedback and dynamic refinement.
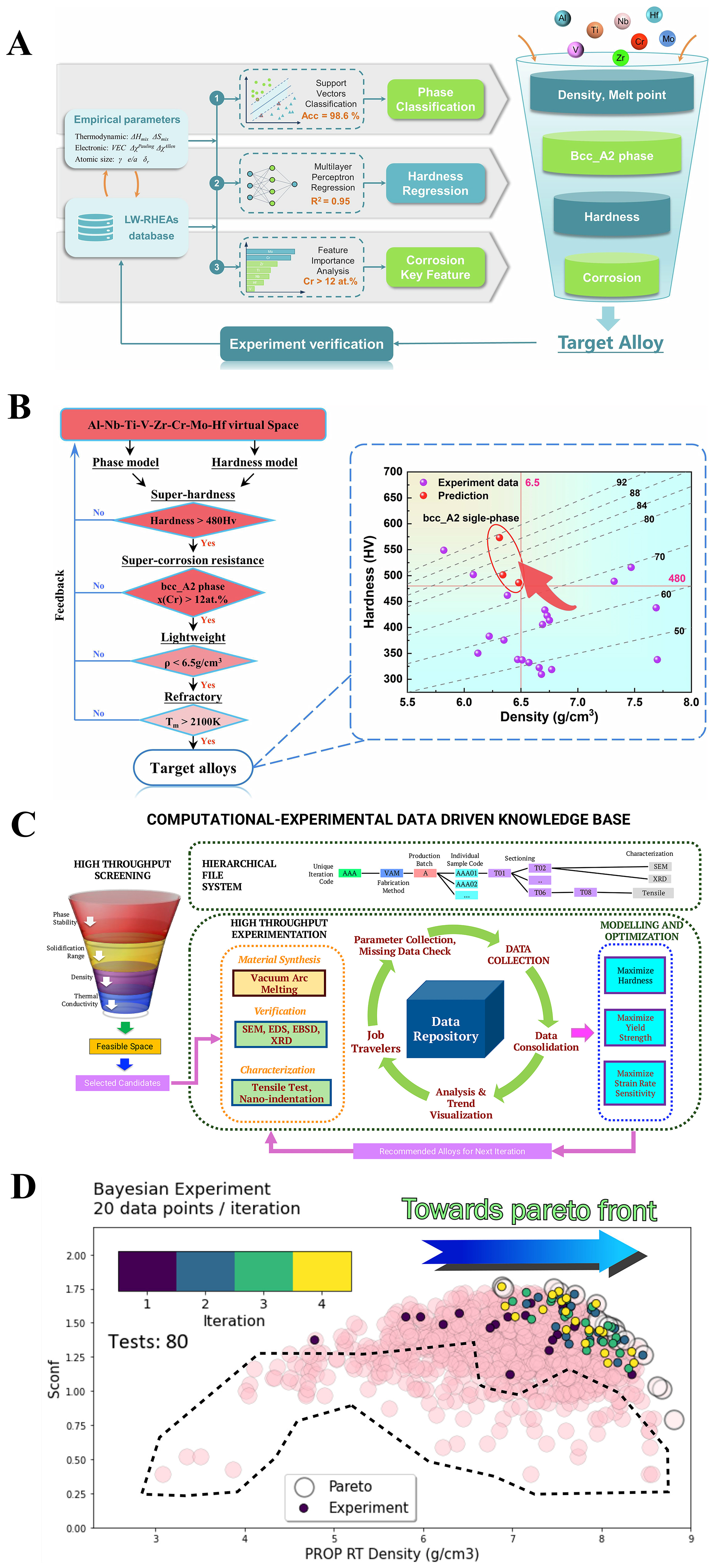
Figure 2. ML-assisted multi-objective optimization of mechanical properties in HEAs. (A) ML-driven multi-objective design strategy for high-performance LW-RHEAs; (B) Sequential filter strategy for multi-objective optimization, resulting in the successful design of three alloys with outstanding comprehensive performance. The figures are quoted with permission from Gao et al.[41]; (C) Schematic illustration of the data management workflow for accelerated materials discovery; (D) Example optimization simulation aimed at maximizing room temperature properties derived from thermodynamic calculations. The figures are quoted with permission from Hastings et al.[42].
Building on this foundation, Hastings et al. proposed a closed-loop Bayesian optimization framework (BIRDSHOT) to accelerate discovery in FCC HEA systems[42], as shown in Figure 2C and D. By integrating high-throughput simulations, phase stability constraints, and batch Bayesian optimization guided by expected hypervolume improvement (EHVI), the authors efficiently explored a high-dimensional Al-V-Cr-Fe-Co-Ni space. Within only five iterative cycles—covering just 0.15% of the design space—they identified multiple non-trivial Pareto-optimal alloys simultaneously optimized for strength, hardness, and strain-rate sensitivity. This approach represents a significant methodological advancement, enabling data-driven exploration with minimal experimental burden and dynamic learning.
To further generalize these frameworks for practical HEA design, an adaptive experiment-design workflow can be conceptualized to connect sequential filtering and Bayesian optimization in a unified loop. A concise recipe is as follows: (1) Define design objectives: separate hard constraints (phase, density, thermal limits) from soft objectives (strength, hardness, corrosion); (2) Assemble an initial dataset: combine literature and prior experiments, ensuring representative sampling of key subspaces; (3) Apply one-shot sequential filtering: reduce the design space using interpretable ML models or physical rules; (4) Launch Bayesian optimization: initialize Gaussian process surrogates with the filtered dataset; (5) Iterate via batch EHVI-guided exploration: suggest 5-10 compositions per cycle, balancing exploration of uncertain regions and exploitation of promising ones; (6) Incorporate feedback and retrain models: update surrogate models with new data until performance metrics converge or Pareto front stabilizes.
Machine learning potentials for accelerated large scale molecular dynamics
Molecular dynamics (MD) is crucial for revealing the structure-property relationships of HEAs, a process that heavily depends on the selected interatomic potential. Conventional MD based on empirical potentials is computationally efficient but often lacks sufficient accuracy[43], while ab initio molecular dynamics offers higher fidelity but is typically more computationally expensive[44]. Machine learning potentials (MLPs), especially high-dimensional neural network potentials (HDNNPs) introduced by Behler and Parrinello[45], bridge this gap by balancing quantum-level accuracy with efficiency. As shown in Figure 3A, HDNNPs express total energy as a sum of atomic contributions based on local environments[46]. Building on the second-generation HDNNP framework[46] [Figure 3B], Fan et al. developed the neuroevolution potential (NEP), which utilizes a single hidden layer feed-forward neural network[47].
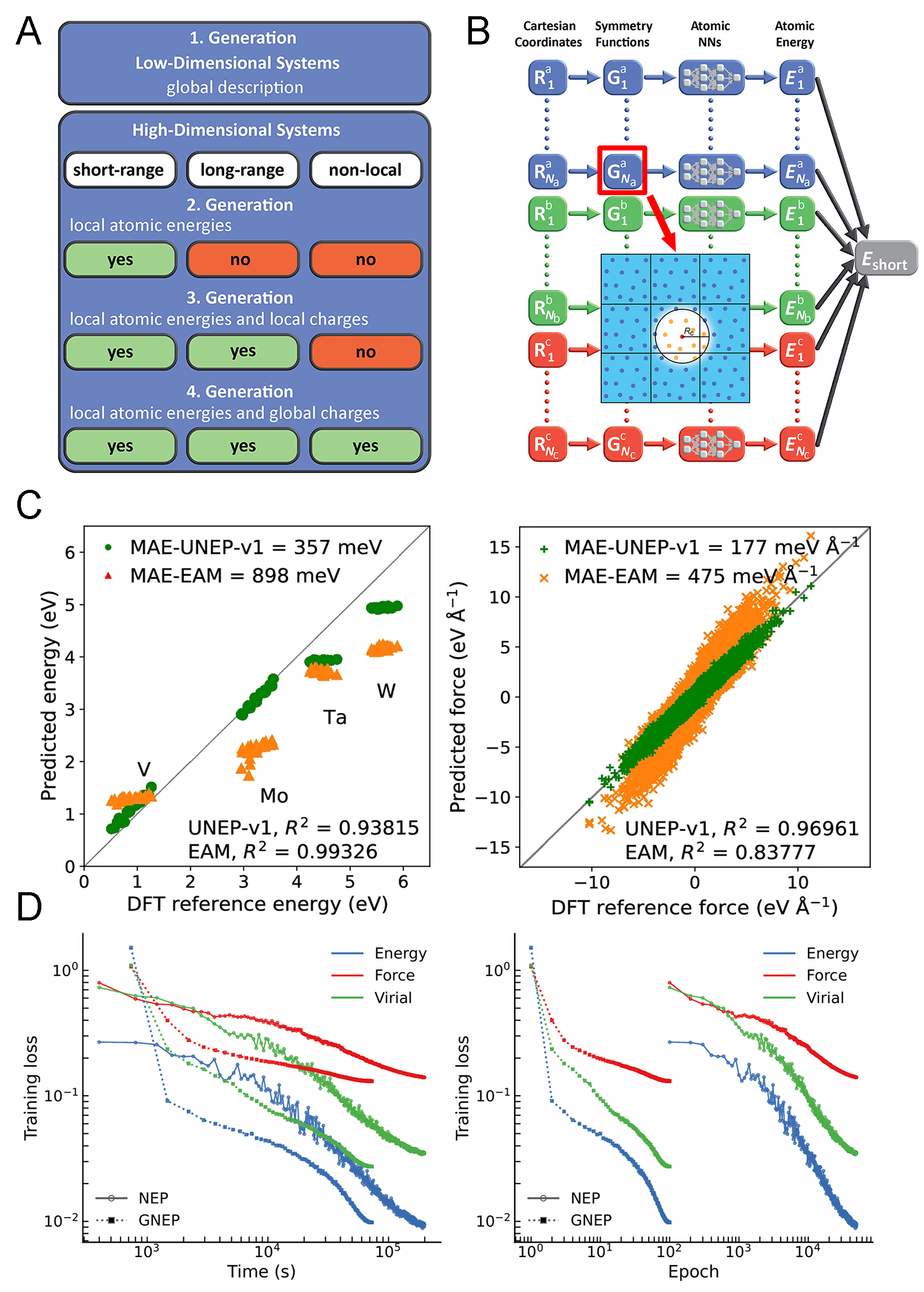
Figure 3. MLP for efficient atomistic simulations of HEAs. (A) Overview of the four generations of NNP; (B) Schematic illustration of the second-generation NNP architecture and its short-range environment descriptors. The figures are quoted with permission from Behler[46]; (C) Performance evaluation of UNEP-v1 for MoTaVW alloys, including mono-vacancy formation energies from UNEP-v1 and EAM, as well as comparisons of UNEP-v1, EAM, and DFT results for equimolar MoTaVW alloys. The figures are quoted with permission from
Extending this work, Song et al. developed a general-purpose machine-learned potential (UNEP-v1) for 16 elemental metals and their alloys[48]. Benchmarking against the embedded-atom method (EAM) potential[49] confirmed its superior accuracy. The model was applied to large-scale MD simulations involving millions of atoms in a BCC MoTaVW HEA system, as shown in Figure 3C. The results demonstrated that UNEP-v1 outperforms EAM potential and is well-suited for investigating structural and mechanical behavior in large-scale atomistic simulations[48].
Building upon these developments, a gradient-optimized NEP (GNEP) was subsequently introduced, in which explicit analytical gradients together with the Adam optimizer were adopted for parameter training[50]. As illustrated in Figure 3D, GNEP was observed to reach low-error plateaus within significantly fewer epochs and reduced wall-clock time compared with the NEP when fitting a Pd-Cu-Ni-P alloy system[50]. This optimization considerably reduces the computational cost of potential construction and is expected to be particularly advantageous for future investigations of HEAs.
CHALLENGES AND FUTURE OUTLOOK
ML is reshaping the traditional trial-and-error paradigm by effectively navigating the vast compositional space to guide experimental discovery. However, significant challenges remain. A major limitation is the scarcity and imbalance of high-quality, labeled data. The lack of standardized databases and the difficulty in integrating diverse data from experiments and simulations hinder the generalizability and robustness of current ML models. Moreover, model interpretability remains limited. Most existing approaches lack physical insight, reducing the trustworthiness when extrapolating beyond known compositional domains. At the multiscale modeling level, MLPs have successfully bridged atomistic and mesoscale simulations with near-DFT accuracy. However, linking these to continuum-level mechanical or thermodynamic descriptions remains challenging due to the intrinsic chemical disorder, sluggish diffusion, and anisotropic deformation mechanisms unique to HEAs.
To overcome these challenges, several key directions may be prioritized. First, building open-access, diverse, and high-fidelity HEA databases that integrate experimental and computational data is critical. Reliable datasets should include not only the precise composition and phase label, but also the processing history, measurement conditions, and associated uncertainties to ensure reproducibility and transferability of models[51]. Several open databases are accessible to HEA-related resources, including AFLOW[52], Materials Project[53], OQMD[54], NOMAD[55] and COD’HEM[56]. Establishing community-wide standards for metadata annotation and data benchmarking will be essential for developing trustworthy ML models and ensuring the comparability of HEA studies across different research groups. Second, physics-informed ML offers a promising path forward by embedding thermodynamic, crystallographic, or mechanistic constraints into model architectures, thus enhancing both extrapolation ability and interpretability. Third, integrating ML with multi-scale modeling frameworks—including CALPHAD, DFT, MD, and finite element methods—will enable comprehensive, scale-bridging insights from atomic structure to macroscopic properties. Finally, coupling ML models with high-throughput experimental platforms and automated synthesis-characterization pipelines could facilitate closed-loop autonomous alloy design systems, substantially accelerating the materials discovery cycle.
Looking forward, the increasing demand for HEAs in lightweight aerospace structures, nuclear radiation-resistant components, and corrosion-resistant extreme-environment systems further underscores the need for data-efficient, physics-informed, uncertainty-aware, and multi-scale ML frameworks capable of accelerating application-driven alloy discovery. These emerging use cases place stringent constraints on mechanical and chemical performance, highlighting the urgency of developing ML approaches that can generalize reliably across vast and sparsely sampled composition spaces.
In summary, ML is poised to become a cornerstone of intelligent HEA discovery. As methods become more data-efficient, interpretable, and seamlessly integrated with experimental workflows, the materials science community can move beyond empirical heuristics toward a new paradigm of predictive and autonomous design. This evolution will not only advance HEA research but also broadly contribute to the realization of the “fourth paradigm” of scientific discovery in materials science.
DECLARATIONS
Authors’ contributions
Data analysis, interpretation and manuscript draft: Yang, N.
Performed data acquisition and collected references: Huang, H.
Provided revision, acquired funding and supervision: Zhou, J.; Sun, Z.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (No. 52332005).
Conflicts of interest
Sun, Z. is Guest Editor of the Special Issue “Machine Learning/AI-Assisted Development of High-Performance Alloys” and Associate Editor of Journal of Materials Informatics. Sun, Z. was not involved in any steps of editorial processing, notably including reviewers selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Cantor, B.; Chang, I.; Knight, P.; Vincent, A. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A. 2004, 375-7, 213-8.
3. Yeh, J. W.; Chen, S. K.; Lin, S. J.; et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299-303.
4. Zhang, Y.; Zuo, T. T.; Tang, Z.; et al. Microstructures and properties of high-entropy alloys. Prog. Mater. Sci. 2014, 61, 1-93.
5. Du, Y.; Hua, D.; Zhou, Q.; et al. Concurrently achieving strength-ductility combination and robust anti-wear performance in an in-situ high-entropy bulk metallic glass composite. Compos. Part. B. Eng. 2024, 272, 111222.
6. Yang, N.; Zhu, L.; Liu, H.; Zhou, J.; Sun, Z. Enhancing the mechanical properties of TiZr-based multi-principal element alloys via leveraging multiple short-range orders: An atomic-scale study. J. Mater. Sci. Technol. 2025, 227, 133-41.
7. El, Atwani. O.; Vo, H. T.; Tunes, M. A.; et al. A quinary WTaCrVHf nanocrystalline refractory high-entropy alloy withholding extreme irradiation environments. Nat. Commun. 2023, 14, 2516.
8. Guo, Y.; Peng, J.; Peng, S.; An, F.; Lu, W.; Li, Z. Improving oxidation resistance of TaMoZrTiAl refractory high entropy alloys via Nb and Si alloying. Corros. Sci. 2023, 223, 111455.
9. Zemanate, A. M.; Jorge Júnior, A. M.; de Lima Andreani, G. F.; Roche, V.; Cardoso, K. R. Corrosion behavior of AlCoCrFeNix high entropy alloys. Electrochim. Acta. 2023, 441, 141844.
10. Panda, P. K.; Liu, K.; Lin, Y.; et al. High-entropy carbon nanodots as metal-free electrochemical catalysts for oxygen reduction and oxygen evolution reactions. Emergent. Mater. 2025, 8, 5591-604.
11. Wang, L.; Ding, J.; Chen, S.; et al. Tailoring planar slip to achieve pure metal-like ductility in body-centred-cubic multi-principal element alloys. Nat. Mater. 2023, 22, 950-7.
12. Feng, R.; Zhang, C.; Gao, M. C.; et al. High-throughput design of high-performance lightweight high-entropy alloys. Nat. Commun. 2021, 12, 4329.
13. Agrawal, A.; Choudhary, A. Perspective: materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL. Mater. 2016, 4, 053208.
14. Himanen, L.; Geurts, A.; Foster, A. S.; Rinke, P. Data-driven materials science: status, challenges, and perspectives. Adv. Sci. 2019, 6, 1900808.
15. Su, Y.; Fu, H.; Bai, Y.; Jiang, X.; Xie, J. Progress in materials genome engineering in China. Acta. Metall. Sin. 2020, 56, 1313-23. (in Chinese).
16. Rao, Z.; Tung, P. Y.; Xie, R.; et al. Machine learning-enabled high-entropy alloy discovery. Science 2022, 378, 78-85.
17. Yuan, J.; Li, Z.; Yang, Y.; et al. Applications of machine learning method in high-performance materials design: a review. J. Mater. Inf. 2024, 4, 14.
18. Wan, X.; Li, Z.; Yu, W.; et al. Machine learning paves the way for high entropy compounds exploration: challenges, progress, and outlook. Adv. Mater. 2025, 37, e2305192.
19. Yan, Y.; Hu, X.; Liao, Y.; Zhou, Y.; He, W.; Zhou, T. Recent machine learning-driven investigations into high entropy alloys: a comprehensive review. J. Alloys. Compd. 2025, 1010, 177823.
20. Ye, Y.; Wang, Q.; Lu, J.; Liu, C.; Yang, Y. High-entropy alloy: challenges and prospects. Mater. Today. 2016, 19, 349-62.
21. Wei, D.; Wang, L.; Zhang, Y.; et al. Metalloid substitution elevates simultaneously the strength and ductility of face-centered-cubic high-entropy alloys. Acta. Mater. 2022, 225, 117571.
22. Chen, S.; Aitken, Z. H.; Pattamatta, S.; et al. Simultaneously enhancing the ultimate strength and ductility of high-entropy alloys via short-range ordering. Nat. Commun. 2021, 12, 4953.
23. Qiu, Y.; Hu, Y.; Taylor, A.; et al. A lightweight single-phase AlTiVCr compositionally complex alloy. Acta. Mater. 2017, 123, 115-24.
24. Murayama, Y.; Hanada, S. High temperature strength, fracture toughness and oxidation resistance of Nb-Si-Al-Ti multiphase alloys. Sci. Technol. Adv. Mater. 2018, 3, 145-56.
25. Liu, X.; Zhang, J.; Pei, Z. Machine learning for high-entropy alloys: Progress, challenges and opportunities. Prog. Mater. Sci. 2023, 131, 101018.
26. Zhao, Y. M.; Zhang, J. Y.; Liaw, P. K.; Yang, T. Machine learning-based computational design methods for high-entropy alloys. High. Entropy. Alloys. Mater. 2025, 3, 41-100.
27. Shimakawa, H.; Kumada, A.; Sato, M. Extrapolative prediction of small-data molecular property using quantum mechanics-assisted machine learning. NPJ. Comput. Mater. 2024, 10, 11.
28. Yang, Z.; Li, S.; Li, S.; Yang, J.; Liu, D. A two-step data augmentation method based on generative adversarial network for hardness prediction of high entropy alloy. Comput. Mater. Sci. 2023, 220, 112064.
29. Sulley, G. A.; Raush, J.; Montemore, M. M.; Hamm, J. Accelerating high-entropy alloy discovery: efficient exploration via active learning. Scr. Mater. 2024, 249, 116180.
30. Guo, S.; Ng, C.; Lu, J.; Liu, C. T. Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J. Appl. Phys. 2011, 109, 103505.
31. Yang, X.; Zhang, Y. Prediction of high-entropy stabilized solid-solution in multi-component alloys. Mater. Chem. Phys. 2012, 132, 233-8.
32. Lv, S.; Li, Q.; Zhang, X.; Jiao, X.; Liu, D. CALPHAD-aided design for superior mechanical behavior in Ti40Zr20Hf40-xCrx eutectic refractory high-entropy alloys. Mater. Charact. 2024, 217, 114393.
33. King, D.; Cheung, S.; Humphry-baker, S.; et al. High temperature, low neutron cross-section high-entropy alloys in the Nb-Ti-V-Zr system. Acta. Mater. 2019, 166, 435-46.
34. Kumar, S.; Linda, A.; Shadangi, Y.; Jindal, V. Influence of micro-segregation on the microstructure, and microhardness of MoNbTaxTi1-xW refractory high entropy alloys: experimental and DFT approach. Intermetallics 2024, 164, 108080.
35. Alkraidi, A. B. N.; Mansoor, M.; Boztemur, B.; et al. Computational alloy design, synthesis, and characterization of WMoNbVCrx refractory high entropy alloy prepared by vacuum arc melting. J. Alloys. Compd. 2024, 1003, 175510.
36. Liu, S.; Bocklund, B.; Diffenderfer, J.; et al. A comparative study of predicting high entropy alloy phase fractions with traditional machine learning and deep neural networks. NPJ. Comput. Mater. 2024, 10, 172.
37. Zhou, Z.; Zhou, Y.; He, Q.; Ding, Z.; Li, F.; Yang, Y. Machine learning guided appraisal and exploration of phase design for high entropy alloys. NPJ. Comput. Mater. 2019, 5, 128.
38. Zhao, S.; Yuan, R.; Liao, W.; et al. Descriptors for phase prediction of high entropy alloys using interpretable machine learning. J. Mater. Chem. A. 2024, 12, 2807-19.
39. Jiang, W.; Wang, X.; Li, S.; Ma, T.; Wang, Y.; Zhu, D. A lightweight Al0.8Nb0.5Ti2V2Zr0.5 refractory high entropy alloy with high specific yield strength. Mater. Lett. 2022, 328, 133144.
40. Jiang, W.; Wang, Y.; Wang, X.; et al. Effect of Al on microstructure and mechanical properties of lightweight AlxNb0.5TiV2Zr0.5 refractory high entropy alloys. Mater. Sci. Eng. A. 2023, 865, 144628.
41. Gao, T.; Gao, J.; Yang, S.; Zhang, L. Data-driven design of novel lightweight refractory high-entropy alloys with superb hardness and corrosion resistance. NPJ. Comput. Mater. 2024, 10, 256.
42. Hastings, T.; Mulukutla, M.; Khatamsaz, D.; et al. Accelerated multi-objective alloy discovery through efficient bayesian methods: application to the FCC high entropy alloy space. Acta. Mater. 2025, 297, 121173.
43. Harrison, J. A.; Schall, J. D.; Maskey, S.; Mikulski, P. T.; Knippenberg, M. T.; Morrow, B. H. Review of force fields and intermolecular potentials used in atomistic computational materials research. Appl. Phys. Rev. 2018, 5, 031104.
44. Li, C.; Voth, G. A. Using machine learning to greatly accelerate path integral ab initio molecular dynamics. J. Chem. Theory. Comput. 2022, 18, 599-604.
45. Behler, J.; Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007, 98, 146401.
46. Behler, J. Four generations of high-dimensional neural network potentials. Chem. Rev. 2021, 121, 10037-72.
47. Fan, Z.; Zeng, Z.; Zhang, C.; et al. Neuroevolution machine learning potentials: Combining high accuracy and low cost in atomistic simulations and application to heat transport. Phys. Rev. B. 2021, 104, 104309.
48. Song, K.; Zhao, R.; Liu, J.; et al. General-purpose machine-learned potential for 16 elemental metals and their alloys. Nat. Commun. 2024, 15, 10208.
49. Zhou, X. W.; Johnson, R. A.; Wadley, H. N. G. Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers. Phys. Rev. B. 2004, 69, 144113.
50. Huang, H.; Peng, J.; Li, K.; Zhou, J.; Sun, Z. Efficient GPU-accelerated training of a neuroevolution potential with analytical gradients. arXiv 2025, arXiv:2507.00528. Available online: https//arxivorg/abs/250700528 (accessed 8 Dec 2025).
51. Ghiringhelli, L. M.; Baldauf, C.; Bereau, T.; et al. Shared metadata for data-centric materials science. Sci. Data. 2023, 10, 626.
52. Curtarolo, S.; Setyawan, W.; Hart, G. L.; et al. AFLOW: an automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 2012, 58, 218-26.
53. Jain, A.; Ong, S. P.; Hautier, G.; et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL. Mater. 2013, 1, 011002.
54. Saal, J. E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 2013, 65, 1501-9.
55. Draxl, C.; Scheffler, M. The NOMAD laboratory: from data sharing to artificial intelligence. J. Phys. Mater. 2019, 2, 036001.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.